
|
Suscribirse a Esclerosis Lateral Amiotrofica - ELA Argentina | |
| Ver archivos en groups.google.com.ar | ||
miércoles, mayo 31, 2006
Algunas Consideraciones sobre la nota de Diario Clarín y otros temas
C.A.L.Mo.
A continuación se transcriben algunos fragmentos de la conferencia brindada por el Dr. Alberto Dubrovsky en la sede de C.A.L.Mo., el día 22 de agosto de 2003.
El Dr Dubrovsky es Profesor Adjunto de Neurología de la UBA y Jefe de la Sección de Enfermedades Neuromusculares del Centro Neurológico del Hospital Francés de Buenos Aires
Tema: Algunas Consideraciones sobre la nota de Diario Clarín y otros temas
(Sábado 09/08/2003)
COMO FUNCIONA EL SISTEMA NEUROMUSCULAR
Los músculos sirven para que nos movamos. Gracias a ellos movilizamos los brazos, las piernas, la lengua, los ojos, párpados y podemos tragar y respirar. Para quienes dudan, los músculos no son otra cosa que la carne, que todos comemos.
El músculo es un tejido constituido como todos los tejidos por células que en este caso son células especiales llamadas fibras musculares que tienen la propiedad de acortarse o sea, contraerse. Para cumplir la función de acortarse y realizar un movimiento, el músculo requiere de que se le envíe una orden. Esa orden llega a través de los nervios y cada nervio está compuesto como un cable con miles de cables más pequeños que inervan cada fibra muscular, para darles la orden de contraerse. Estos cables son en realidad la prolongación de las neuronas que están en la médula espinal.
La neurona es una célula con una capacidad única de generar impulsos de corriente. Estas prolongaciones se llaman "axónes". La neurona tiene la posibilidad de excitarse, desde el punto de vista bio-eléctrico, generando un impulso que se propaga a través del axón y se transmite, ya sea a otra neurona o, en el caso que nos interesa a nosotros, a los músculos. Los nervios están constituidos por miles de axones de otras tantas neuronas. Estas funcionan en forma sincronizada y cuando reciben una orden del cerebro, transmiten sus impulsos al músculo y éste se acorta.
Los cuerpos de las neuronas motoras se encuentran en la médula espinal, que está contenida en la columna vertebral. A ella llegan las órdenes de las neuronas del cerebro para realizar los movimientos
QUE SON LAS ENFERMEDADES DE LA MOTONEURONA
Las enfermedades de la neurona motora producen degeneración de las neuronas que están ubicadas a lo largo de la médula espinal. Entre esas neuronas están las encargadas de distintos grupos musculares (como dijimos antes, los responsables del movimiento de los brazos, ojos, piernas, etc.) La enfermedad, entonces, está localizada en las neuronas encargadas del movimiento.
Existen otras neuronas encargadas de la sensibilidad, las que en realidad no envían ordenes de movimiento sino que reciben información sensitiva, por ejemplo: de la piel. Cuando se hacen unas cosquillas o un pinchazo, esa información viaja por las neuronas sensitivas. Pero estas neuronas no están afectadas en este tipo de enfermedades.
A medida que van degenerando (enfermando) las neuronas se pierde el volumen muscular (atrofia). Eventualmente, lo que ocurre es un proceso de compensación parcial, en donde la neurona que está sana es capaz de tomar a su cargo las fibras musculares que quedaron huérfanas, hasta un cierto límite. Pero muchas desaparecen y este proceso sigue así hasta que esta neurona es la que degenera, perdiendo más cantidad de fibras musculares.
Este proceso es de pérdida o "denervación" (porque las fibras musculares se denervan). La velocidad con que este proceso ocurre, tiene que ver con la velocidad de deterioro que produce la enfermedad.
Dependiendo del músculo afectado, será donde va a aparecer primero la sintomatología de la enfermedad. Si la enfermedad empieza a atacar primero a las neuronas encargadas de inervar los músculos de la pierna, la debilidad y la atrofia muscular aparecerá primero en la pierna. En otros casos, aparecerá en los brazos u otros empezarán por tener problemas para tragar o para hablar porque lo que hizo la enfermedad fue atacar las neuronas que inervan los músculos de la deglución o de la fonación.
CUAL ES LA CAUSA
La causa origen o etiología de la enfermedad es en realidad desconocida. No se sabe por que las neuronas motoras inician el proceso de degeneración.
Tampoco se sabe aún por qué la enfermedad empieza en un grupo de neuronas específico en algunos pacientes y en otro grupo distinto, en otros pacientes.. En última instancia, no se sabe cuál es la PRIMERA CAUSA. Se está invirtiendo mucho en investigación desde hace muchos años y en forma intensiva, pero todavía no se conoce la CAUSA. Si en cambio se están empezando a conocer MECANISMOS tal como el del Glutamato que explicaremos luego.
Existen otras hipótesis sobre posibles mecanismos que apuntan a distintas causas.
Se están investigando los llamados factores de riesgo.
Hubo, recientemente una estadística en Italia, que sugirió que un porcentaje mayor de enfermos de E.L.A. se encontraba presente en pacientes deportistas o futbolistas.. También se dijo en algún momento, que era mayor la incidencia de la enfermedad en pobladores rurales. Con posterioridad, esta estadística fue desmentida. La que hace alusión a los futbolistas, se está estudiando. Lo que estas situaciones pueden indicar es la existencia de FACTORES DE RIESGO. O sea que existirían ciertas actividades, lugares de vivienda, profesiones, contactos con mascotas, hábitos, enfermedades concomitantes o condiciones que, posiblemente al actuar con otros factores ayuden a desencadenar la enfermedad pero no deben confundirse con la CAUSA. Se podría decir que son “pistas”que pueden ser útiles para comenzar un camino de investigación hacia la causa. Es importante entonces que estos factores de riesgo se deben poder comprobar mediante estadísticas y validaciones matemáticas y no por el hallazgo de meras coincidencias ocasionales o suposiciones o trabajos carentes de rigor científico.
CONSIDERACIONES SOBRE LA NOTA DEL DIARIO CLARIN
Ver nota en: Hallan el gen de una rara enfermedad
Entre un 5 a un 10% de los casos de E.L.A., son formas FAMILIARES o HEREDITARIAS de la enfermedad. Esto quiere decir que, aproximadamente el 95% de los casos NO son hereditarios.
Dentro de este 5%, el 20% de estos casos, se deben a mutaciones de un gen (SOD1) ubicado en el cromosoma 21. Hoy se sabe que no es solamente en el cromosoma 21, hay muchas otras localizaciones, como el cromosoma 9, el 13, etc. En este caso se conoce la CAUSA, se sabe que hay una alteración en ese gen, pero no se sabe lo que esa alteración termina produciendo para que al final se muera la moto neurona.
En el 95% de los casos restantes, llamadas ESPORÁDICAS, que es la forma más habitual y común de la enfermedad, no hay un gen identificado que produzca la enfermedad.
Lo que se encontró, buscando "a ciegas", es que existe un factor vascular determinado genéticamente, que tiene que ver con el crecimiento de los vasos, que está alterado en un alto porcentaje de pacientes con E.L.A.
DE ALLI A QUE ESE GEN SEA EL CAUSANTE DE LA E.L.A. ESTÁ MUY LEJOS DE SER PROBADO.
Puede ser un epi -fenómeno, puede ser algo que venga adjunto, puede ser que no tenga nada que ver y que ese gen esté alterado también en pacientes con cualquier otra enfermedad. En todo caso podría considerárselo por ahora un “factor de riesgo mas”
Es una revelación interesante, porque por primera vez se encuentra una alteración genética (aunque no necesariamente causante) en las formas esporádicas de la enfermedad.
¿Qué se hace con esta información?
Todavía hay mucho camino por recorrer para ver que valor pueda tener este hallazgo. El título de la nota aparecida en el diario Clarín lleva a la confusión.
Una nota de prensa de la ALS Asociation, la Asociación americana de E.L.A., pone en el contexto apropiado lo comentado por el diario Clarín:
-"El factor de crecimiento vascular alterado podría ser un factor de riesgo, o sea que podría colocar al sujeto en riesgo de padecer la enfermedad porque estos hechos, pareciera ser que vienen juntos.” Pero no más que un "parecería ser un marcador de factor de riesgo". De ninguna manera está probado que este gen cause la enfermedad. " –
GENES , ELA, GLUTAMATO Y OTRAS CUESTIONES
Un GEN es un pequeño segmento ADN, que contiene un código a través del cual le "dice a la célula" que proteína tiene que fabricar. Todas las células, para sobrevivir y nutrirse necesitan fabricar sustancias. Dentro del núcleo celular se encuentra el ADN.
Cada una de las células de nuestro organismo: de la piel, del hígado, músculos, etc. tiene la misma información genética. Los genes están contenidos en los cromosomas. Una forma hereditaria de la enfermedad está causada por una alteración de uno de esos genes (alteración = mutación = cambio) que codifica (en otras palabras contiene las instrucciones para que la célula fabrique) la enzima SOD1 que es una proteína que tiene una determinada función dentro de la neurona. Cuando esta sustancia, la SOD1, se altera, produce la muerte de la neurona. En este caso se encontró la causa y se sabe el resultado final, pero entre medio, suceden muchos eventos que no se conocen con exactitud. En las formas no hereditarias de la ELA que llamamos esporádicas no se conoce la causa aunque si se conocen algunos mecanismos; uno de ellos involucra al Glutamato.
Las células nerviosas se comunican entre sí mediante las SINAPSIS (punto de contacto entre las neuronas) Cada célula recibe miles de contactos o sinapsis con informaciones de otras tantas neuronas y decide si manda o no el estímulo. Las señales entre células nerviosas se transmiten a nivel de las sinapsis por estímulos químicos o mediadores químicos. Las células liberan en las sinapsis, substancias que son captadas por la otra célula. Esas substancias son los neurotransmisores. Uno de esos neurotransmisores es el glutamato.
Lo que se ha probado es que en esta enfermedad, las neuronas motoras se vuelven particularmente sensibles al glutamato y éste las daña hasta matarlas. No se sabe qué es lo que las vuelve sensibles al glutamato. Una teoría dice que el glutamato se acumula porque falla un mecanismo que permite que cuando se libera el glutamato, el exceso del mismo es eliminado por un sistema de transporte que se lo lleva. Este sistema podría fallar a porque fallaría un gen , que no fabrica la proteína encargada de llevarse el glutamato. Todas estas son hipótesis que, posiblemente sean parte de la cadena de eventos que desencadenan la enfermedad.
En la medida en que se conozcan las causas, más herramientas se van a tener para una terapéutica racional, dirigida a combatir la causa y no sólo el mecanismo.
COMO SE APRUEBAN MEDICAMENTOS
RILUTEK y OTRAS DROGAS
Lo que hace el RILUTEK es interferir en el mecanismo del glutamato y frenar la evolución de la enfermedad. El RILUTEK no cura la enfermedad sino que la retarda.
La EVIDENCIA CIENTÍFICA está basada en conocimientos científicos y debe demostrar a través de estadísticas que los resultados obtenidos no se dan por azar.
Cuando surgió el tema del glutamato, se decidió buscar drogas que frenaran la liberación del glutamato o la captación del mismo por parte de la neurona.
Así se decidió estudiar el Riluzole que es un compuesto que tiene precisamente esas propiedades.
Es interesante que la gente tenga idea del proceso que lleva la investigación de una nueva droga desde que el laboratorio adquiere los derechos de esa sustancia o molécula hasta que puede ser vendida en las farmacias.
En una PRIMERA FASE, la droga se prueba en ratones y se verifica que estos no mueran como consecuencia de su uso. De ser así, se desecha la molécula. Una vez superada la primera fase, se prueban dosis máximas de droga, que puedan soportar los ratones hasta su muerte. Se establece así la dosis y los efectos producidos en los ratones. También se estudia qué es lo que la droga produce en el animal de experimentación, a través de múltiples y complicados estudios. Luego se pasa a la FASE HUMANA, en donde a sujetos sanos se les suministra la droga y se estudian los efectos secundarios (se trata de voluntarios pagos).
Luego de varias etapas se prueba el compuesto en pacientes con la enfermedad para la cual la droga esta destinada comparándose el efecto que produce su uso en pacientes que la toman con aquellos que no la toman. Si las diferencias son muy pequeñas , no se puede saber con certeza que se deban a la droga en estudio y se necesitan para eso más pacientes. A medida que la muestra es más grande (cantidad de pacientes), menor es la posibilidad de error.
Los pacientes se someten a estrictos controles y estos estudios (llamados ensayos terapéuticos) son monitoreados por agencias de salud oficiales (como el ANMAT en la Argentina o la FDA en EEUU)
Los datos de los estudios, los efectos de las drogas en los pacientes, los datos de los exámenes de laboratorio etc etc etc son sometidos a análisis estadísticos que determinaran la utilidad o no de la droga para la enfermedad en cuestión.
Desde el momento en que un laboratorio compra o patenta una molécula como propia, hasta que llega el medicamento a la farmacia, pasan un promedio de 10 años, con una inversión de entre 700 y 800 millones de U$S.
Esta inversión es realizada por el laboratorio que investiga el medicamento y por supuesto debe recuperarla (de allí el costo a veces elevado de muchos medicamentos). También intervienen por supuesto cuestiones de mercado………
Los laboratorios patentan o compran la patente de aquellas moléculas que se supone que puedan llegar a ser comercializadas en un futuro y pocas son las que llegan a esa instancia ya que la mayoría queda en el camino…. El valor final del remedio tiene que ver con el costo del desarrollo de la droga, desde la compra o patentamiento hasta que llega al mostrador.
El RILUTEK, es una droga de investigación que sufrió este proceso.
La E.L.A. como ya vimos tiene a la muerte de las neuronas motoras como común denominador, pero se expresa de maneras distintas en cada sujeto (en cuanto al inicio y progreso de la misma), lo cual hace que la comparación entre grupos de pacientes se deba hacer con grandes cantidades de sujetos, porque si no, no se puede evaluar la evolución de la droga, a menos que el cambio sea drástico (por Ej. la curación de la enfermedad). Con RILUTEK hubo dos estudios que involucraron más de 1000 pacientes, que demostraron que el promedio de los pacientes que tomaron la medicación tuvieron mejor evolución. que aquellos pacientes que no tomaron la medicación..
Hay otras drogas en periodo de prueba tales como la MINOCICLINA , CELECOXIB, FACTORES TROFICOS, CREATINA, PROGESTERONA, ANTIOXIDANTES, TAMOXIFENO, INDINAVIR, COENZIMA Q 10, TOPIRAMATO, ETC.
CONSIDERACIONES FINALES
La enfermedad se manifiesta por atrofia muscular y pérdida de la fuerza.
Es importante saber que se pueden perder del 30 al 40% de las neuronas motoras sin tener síntomas de debilidad. Por eso es muy difícil saber cuando empezó la enfermedad. Cuando el paciente llega a la consulta, tiene ya una perdida importante de neuronas motoras.
En el curso evolutivo de la enfermedad sin embargo aparecen muchos otros síntomas que requieren control por parte del medico.
La enfermedad es, hasta hoy, incurable. Pero no es intratable. Con frecuencia estos términos se confunden. Además de la medicación destinada al tratamiento especifico de la enfermedad destinada a disminuir su progresión se debe poner énfasis en otros aspectos del tratamiento.
TRATAR es mejorar la calidad de vida y brindar alivio de síntomas y complicaciones.
Existen medicaciones y procedimientos para aliviar muchos de los padecimientos por los que se ve aquejado el paciente con ELA como los calambres, espasticidad, dificultades deglutorias, exceso de saliva, etc.
El medico debe intervenir para aliviar la depresión y la ansiedad, para mejorar la nutrición, tratar los trastornos respiratorios, aconsejar en el uso de férulas u ortesis y del tratamiento kinesiológico, ayudar con los problemas de comunicación, etc.
No existen esquemas fijos y el manejo de cada paciente es individual y debe ser diseñado en cada caso por el medico tratante.
Es importante que el profesional a cargo conozca la enfermedad y sepa que esperar de la misma según la evolución de cada paciente para así poder intervenir apropiadamente.
Fuente:
http://www.calmo.org.ar
A continuación se transcriben algunos fragmentos de la conferencia brindada por el Dr. Alberto Dubrovsky en la sede de C.A.L.Mo., el día 22 de agosto de 2003.
El Dr Dubrovsky es Profesor Adjunto de Neurología de la UBA y Jefe de la Sección de Enfermedades Neuromusculares del Centro Neurológico del Hospital Francés de Buenos Aires
Tema: Algunas Consideraciones sobre la nota de Diario Clarín y otros temas
(Sábado 09/08/2003)
COMO FUNCIONA EL SISTEMA NEUROMUSCULAR
Los músculos sirven para que nos movamos. Gracias a ellos movilizamos los brazos, las piernas, la lengua, los ojos, párpados y podemos tragar y respirar. Para quienes dudan, los músculos no son otra cosa que la carne, que todos comemos.
El músculo es un tejido constituido como todos los tejidos por células que en este caso son células especiales llamadas fibras musculares que tienen la propiedad de acortarse o sea, contraerse. Para cumplir la función de acortarse y realizar un movimiento, el músculo requiere de que se le envíe una orden. Esa orden llega a través de los nervios y cada nervio está compuesto como un cable con miles de cables más pequeños que inervan cada fibra muscular, para darles la orden de contraerse. Estos cables son en realidad la prolongación de las neuronas que están en la médula espinal.
La neurona es una célula con una capacidad única de generar impulsos de corriente. Estas prolongaciones se llaman "axónes". La neurona tiene la posibilidad de excitarse, desde el punto de vista bio-eléctrico, generando un impulso que se propaga a través del axón y se transmite, ya sea a otra neurona o, en el caso que nos interesa a nosotros, a los músculos. Los nervios están constituidos por miles de axones de otras tantas neuronas. Estas funcionan en forma sincronizada y cuando reciben una orden del cerebro, transmiten sus impulsos al músculo y éste se acorta.
Los cuerpos de las neuronas motoras se encuentran en la médula espinal, que está contenida en la columna vertebral. A ella llegan las órdenes de las neuronas del cerebro para realizar los movimientos
QUE SON LAS ENFERMEDADES DE LA MOTONEURONA
Las enfermedades de la neurona motora producen degeneración de las neuronas que están ubicadas a lo largo de la médula espinal. Entre esas neuronas están las encargadas de distintos grupos musculares (como dijimos antes, los responsables del movimiento de los brazos, ojos, piernas, etc.) La enfermedad, entonces, está localizada en las neuronas encargadas del movimiento.
Existen otras neuronas encargadas de la sensibilidad, las que en realidad no envían ordenes de movimiento sino que reciben información sensitiva, por ejemplo: de la piel. Cuando se hacen unas cosquillas o un pinchazo, esa información viaja por las neuronas sensitivas. Pero estas neuronas no están afectadas en este tipo de enfermedades.
A medida que van degenerando (enfermando) las neuronas se pierde el volumen muscular (atrofia). Eventualmente, lo que ocurre es un proceso de compensación parcial, en donde la neurona que está sana es capaz de tomar a su cargo las fibras musculares que quedaron huérfanas, hasta un cierto límite. Pero muchas desaparecen y este proceso sigue así hasta que esta neurona es la que degenera, perdiendo más cantidad de fibras musculares.
Este proceso es de pérdida o "denervación" (porque las fibras musculares se denervan). La velocidad con que este proceso ocurre, tiene que ver con la velocidad de deterioro que produce la enfermedad.
Dependiendo del músculo afectado, será donde va a aparecer primero la sintomatología de la enfermedad. Si la enfermedad empieza a atacar primero a las neuronas encargadas de inervar los músculos de la pierna, la debilidad y la atrofia muscular aparecerá primero en la pierna. En otros casos, aparecerá en los brazos u otros empezarán por tener problemas para tragar o para hablar porque lo que hizo la enfermedad fue atacar las neuronas que inervan los músculos de la deglución o de la fonación.
CUAL ES LA CAUSA
La causa origen o etiología de la enfermedad es en realidad desconocida. No se sabe por que las neuronas motoras inician el proceso de degeneración.
Tampoco se sabe aún por qué la enfermedad empieza en un grupo de neuronas específico en algunos pacientes y en otro grupo distinto, en otros pacientes.. En última instancia, no se sabe cuál es la PRIMERA CAUSA. Se está invirtiendo mucho en investigación desde hace muchos años y en forma intensiva, pero todavía no se conoce la CAUSA. Si en cambio se están empezando a conocer MECANISMOS tal como el del Glutamato que explicaremos luego.
Existen otras hipótesis sobre posibles mecanismos que apuntan a distintas causas.
Se están investigando los llamados factores de riesgo.
Hubo, recientemente una estadística en Italia, que sugirió que un porcentaje mayor de enfermos de E.L.A. se encontraba presente en pacientes deportistas o futbolistas.. También se dijo en algún momento, que era mayor la incidencia de la enfermedad en pobladores rurales. Con posterioridad, esta estadística fue desmentida. La que hace alusión a los futbolistas, se está estudiando. Lo que estas situaciones pueden indicar es la existencia de FACTORES DE RIESGO. O sea que existirían ciertas actividades, lugares de vivienda, profesiones, contactos con mascotas, hábitos, enfermedades concomitantes o condiciones que, posiblemente al actuar con otros factores ayuden a desencadenar la enfermedad pero no deben confundirse con la CAUSA. Se podría decir que son “pistas”que pueden ser útiles para comenzar un camino de investigación hacia la causa. Es importante entonces que estos factores de riesgo se deben poder comprobar mediante estadísticas y validaciones matemáticas y no por el hallazgo de meras coincidencias ocasionales o suposiciones o trabajos carentes de rigor científico.
CONSIDERACIONES SOBRE LA NOTA DEL DIARIO CLARIN
Ver nota en: Hallan el gen de una rara enfermedad
Entre un 5 a un 10% de los casos de E.L.A., son formas FAMILIARES o HEREDITARIAS de la enfermedad. Esto quiere decir que, aproximadamente el 95% de los casos NO son hereditarios.
Dentro de este 5%, el 20% de estos casos, se deben a mutaciones de un gen (SOD1) ubicado en el cromosoma 21. Hoy se sabe que no es solamente en el cromosoma 21, hay muchas otras localizaciones, como el cromosoma 9, el 13, etc. En este caso se conoce la CAUSA, se sabe que hay una alteración en ese gen, pero no se sabe lo que esa alteración termina produciendo para que al final se muera la moto neurona.
En el 95% de los casos restantes, llamadas ESPORÁDICAS, que es la forma más habitual y común de la enfermedad, no hay un gen identificado que produzca la enfermedad.
Lo que se encontró, buscando "a ciegas", es que existe un factor vascular determinado genéticamente, que tiene que ver con el crecimiento de los vasos, que está alterado en un alto porcentaje de pacientes con E.L.A.
DE ALLI A QUE ESE GEN SEA EL CAUSANTE DE LA E.L.A. ESTÁ MUY LEJOS DE SER PROBADO.
Puede ser un epi -fenómeno, puede ser algo que venga adjunto, puede ser que no tenga nada que ver y que ese gen esté alterado también en pacientes con cualquier otra enfermedad. En todo caso podría considerárselo por ahora un “factor de riesgo mas”
Es una revelación interesante, porque por primera vez se encuentra una alteración genética (aunque no necesariamente causante) en las formas esporádicas de la enfermedad.
¿Qué se hace con esta información?
Todavía hay mucho camino por recorrer para ver que valor pueda tener este hallazgo. El título de la nota aparecida en el diario Clarín lleva a la confusión.
Una nota de prensa de la ALS Asociation, la Asociación americana de E.L.A., pone en el contexto apropiado lo comentado por el diario Clarín:
-"El factor de crecimiento vascular alterado podría ser un factor de riesgo, o sea que podría colocar al sujeto en riesgo de padecer la enfermedad porque estos hechos, pareciera ser que vienen juntos.” Pero no más que un "parecería ser un marcador de factor de riesgo". De ninguna manera está probado que este gen cause la enfermedad. " –
GENES , ELA, GLUTAMATO Y OTRAS CUESTIONES
Un GEN es un pequeño segmento ADN, que contiene un código a través del cual le "dice a la célula" que proteína tiene que fabricar. Todas las células, para sobrevivir y nutrirse necesitan fabricar sustancias. Dentro del núcleo celular se encuentra el ADN.
Cada una de las células de nuestro organismo: de la piel, del hígado, músculos, etc. tiene la misma información genética. Los genes están contenidos en los cromosomas. Una forma hereditaria de la enfermedad está causada por una alteración de uno de esos genes (alteración = mutación = cambio) que codifica (en otras palabras contiene las instrucciones para que la célula fabrique) la enzima SOD1 que es una proteína que tiene una determinada función dentro de la neurona. Cuando esta sustancia, la SOD1, se altera, produce la muerte de la neurona. En este caso se encontró la causa y se sabe el resultado final, pero entre medio, suceden muchos eventos que no se conocen con exactitud. En las formas no hereditarias de la ELA que llamamos esporádicas no se conoce la causa aunque si se conocen algunos mecanismos; uno de ellos involucra al Glutamato.
Las células nerviosas se comunican entre sí mediante las SINAPSIS (punto de contacto entre las neuronas) Cada célula recibe miles de contactos o sinapsis con informaciones de otras tantas neuronas y decide si manda o no el estímulo. Las señales entre células nerviosas se transmiten a nivel de las sinapsis por estímulos químicos o mediadores químicos. Las células liberan en las sinapsis, substancias que son captadas por la otra célula. Esas substancias son los neurotransmisores. Uno de esos neurotransmisores es el glutamato.
Lo que se ha probado es que en esta enfermedad, las neuronas motoras se vuelven particularmente sensibles al glutamato y éste las daña hasta matarlas. No se sabe qué es lo que las vuelve sensibles al glutamato. Una teoría dice que el glutamato se acumula porque falla un mecanismo que permite que cuando se libera el glutamato, el exceso del mismo es eliminado por un sistema de transporte que se lo lleva. Este sistema podría fallar a porque fallaría un gen , que no fabrica la proteína encargada de llevarse el glutamato. Todas estas son hipótesis que, posiblemente sean parte de la cadena de eventos que desencadenan la enfermedad.
En la medida en que se conozcan las causas, más herramientas se van a tener para una terapéutica racional, dirigida a combatir la causa y no sólo el mecanismo.
COMO SE APRUEBAN MEDICAMENTOS
RILUTEK y OTRAS DROGAS
Lo que hace el RILUTEK es interferir en el mecanismo del glutamato y frenar la evolución de la enfermedad. El RILUTEK no cura la enfermedad sino que la retarda.
La EVIDENCIA CIENTÍFICA está basada en conocimientos científicos y debe demostrar a través de estadísticas que los resultados obtenidos no se dan por azar.
Cuando surgió el tema del glutamato, se decidió buscar drogas que frenaran la liberación del glutamato o la captación del mismo por parte de la neurona.
Así se decidió estudiar el Riluzole que es un compuesto que tiene precisamente esas propiedades.
Es interesante que la gente tenga idea del proceso que lleva la investigación de una nueva droga desde que el laboratorio adquiere los derechos de esa sustancia o molécula hasta que puede ser vendida en las farmacias.
En una PRIMERA FASE, la droga se prueba en ratones y se verifica que estos no mueran como consecuencia de su uso. De ser así, se desecha la molécula. Una vez superada la primera fase, se prueban dosis máximas de droga, que puedan soportar los ratones hasta su muerte. Se establece así la dosis y los efectos producidos en los ratones. También se estudia qué es lo que la droga produce en el animal de experimentación, a través de múltiples y complicados estudios. Luego se pasa a la FASE HUMANA, en donde a sujetos sanos se les suministra la droga y se estudian los efectos secundarios (se trata de voluntarios pagos).
Luego de varias etapas se prueba el compuesto en pacientes con la enfermedad para la cual la droga esta destinada comparándose el efecto que produce su uso en pacientes que la toman con aquellos que no la toman. Si las diferencias son muy pequeñas , no se puede saber con certeza que se deban a la droga en estudio y se necesitan para eso más pacientes. A medida que la muestra es más grande (cantidad de pacientes), menor es la posibilidad de error.
Los pacientes se someten a estrictos controles y estos estudios (llamados ensayos terapéuticos) son monitoreados por agencias de salud oficiales (como el ANMAT en la Argentina o la FDA en EEUU)
Los datos de los estudios, los efectos de las drogas en los pacientes, los datos de los exámenes de laboratorio etc etc etc son sometidos a análisis estadísticos que determinaran la utilidad o no de la droga para la enfermedad en cuestión.
Desde el momento en que un laboratorio compra o patenta una molécula como propia, hasta que llega el medicamento a la farmacia, pasan un promedio de 10 años, con una inversión de entre 700 y 800 millones de U$S.
Esta inversión es realizada por el laboratorio que investiga el medicamento y por supuesto debe recuperarla (de allí el costo a veces elevado de muchos medicamentos). También intervienen por supuesto cuestiones de mercado………
Los laboratorios patentan o compran la patente de aquellas moléculas que se supone que puedan llegar a ser comercializadas en un futuro y pocas son las que llegan a esa instancia ya que la mayoría queda en el camino…. El valor final del remedio tiene que ver con el costo del desarrollo de la droga, desde la compra o patentamiento hasta que llega al mostrador.
El RILUTEK, es una droga de investigación que sufrió este proceso.
La E.L.A. como ya vimos tiene a la muerte de las neuronas motoras como común denominador, pero se expresa de maneras distintas en cada sujeto (en cuanto al inicio y progreso de la misma), lo cual hace que la comparación entre grupos de pacientes se deba hacer con grandes cantidades de sujetos, porque si no, no se puede evaluar la evolución de la droga, a menos que el cambio sea drástico (por Ej. la curación de la enfermedad). Con RILUTEK hubo dos estudios que involucraron más de 1000 pacientes, que demostraron que el promedio de los pacientes que tomaron la medicación tuvieron mejor evolución. que aquellos pacientes que no tomaron la medicación..
Hay otras drogas en periodo de prueba tales como la MINOCICLINA , CELECOXIB, FACTORES TROFICOS, CREATINA, PROGESTERONA, ANTIOXIDANTES, TAMOXIFENO, INDINAVIR, COENZIMA Q 10, TOPIRAMATO, ETC.
CONSIDERACIONES FINALES
La enfermedad se manifiesta por atrofia muscular y pérdida de la fuerza.
Es importante saber que se pueden perder del 30 al 40% de las neuronas motoras sin tener síntomas de debilidad. Por eso es muy difícil saber cuando empezó la enfermedad. Cuando el paciente llega a la consulta, tiene ya una perdida importante de neuronas motoras.
En el curso evolutivo de la enfermedad sin embargo aparecen muchos otros síntomas que requieren control por parte del medico.
La enfermedad es, hasta hoy, incurable. Pero no es intratable. Con frecuencia estos términos se confunden. Además de la medicación destinada al tratamiento especifico de la enfermedad destinada a disminuir su progresión se debe poner énfasis en otros aspectos del tratamiento.
TRATAR es mejorar la calidad de vida y brindar alivio de síntomas y complicaciones.
Existen medicaciones y procedimientos para aliviar muchos de los padecimientos por los que se ve aquejado el paciente con ELA como los calambres, espasticidad, dificultades deglutorias, exceso de saliva, etc.
El medico debe intervenir para aliviar la depresión y la ansiedad, para mejorar la nutrición, tratar los trastornos respiratorios, aconsejar en el uso de férulas u ortesis y del tratamiento kinesiológico, ayudar con los problemas de comunicación, etc.
No existen esquemas fijos y el manejo de cada paciente es individual y debe ser diseñado en cada caso por el medico tratante.
Es importante que el profesional a cargo conozca la enfermedad y sepa que esperar de la misma según la evolución de cada paciente para así poder intervenir apropiadamente.
Fuente:
http://www.calmo.org.ar
martes, mayo 30, 2006
Algunas consideraciones y actualizaciones sobre el tratamiento de la ELA
Dra. Laura Pirra (Médica neuróloga. Sección de Enfermedades Neuromusculares- servicio de Neurología- Centro Neurológico del Hospital Francés de Buenos Aires)
Tema: Algunas consideraciones y actualizaciones sobre el tratamiento de la ELA
GENERALIDADES:
Para realizar un movimiento, como mover un brazo, caminar, tragar o respirar, entre otros, el cerebro debe enviar una orden hacia los músculos involucrados con esa actividad. Para ello cuenta con un grupo de células nerviosas ubicadas en el cerebro, llamadas Neuronas Motoras Superiores, las que constituyen el primer nivel en el circuito motor, por lo que también se las denomina Neuronas Motoras Primarias. Estas Neuronas motoras primarias se conectan y envían señales, a través de un mecanismo denominado Sinapsis, a otras neuronas ubicadas en la medula espinal, la cual se encuentra dentro de la columna vertebral, a las que llamamos Neuronas Motoras Inferiores o Neuronas motoras secundarias. Estas últimas Neuronas establecen contacto con los músculos a través de largas prolongaciones que se reúnen formando los nervios periféricos. Es decir que la orden se origina en el cerebro, viaja por la médula y los nervios periféricos para luego arribar a los músculos que ejecutarán la orden original.
TIPOS DE ENFERMEDAD DE NEURONA MOTORA:
Las enfermedades de la Neurona Motora pueden afectar exclusivamente a las neuronas Motoras Superiores (ubicadas en el cerebro), como es el caso de la Esclerosis lateral primaria (ELP), o selectivamente a las Neuronas Motoras Inferiores (ubicadas en la medula espinal), como por ejemplo las Atrofias espinales, o pueden afectar ambos grupos de neuronas, Superiores e Inferiores, como en el caso de la Esclerosis Lateral Amiotrófica (ELA)
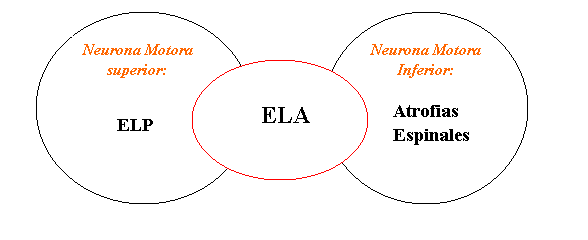 Existen otros tipos de neuronas en el cerebro y la medula espinal, como las que movilizan los ojos, las que transportan la sensibilidad o las que procesan la memoria, las que no son afectadas en este grupo de enfermedades.
Existen otros tipos de neuronas en el cerebro y la medula espinal, como las que movilizan los ojos, las que transportan la sensibilidad o las que procesan la memoria, las que no son afectadas en este grupo de enfermedades.
SINTOMAS EN LA ELA:
Los síntomas van apareciendo gradualmente a medida que se enferman las neuronas motoras, entre ellos podemos mencionar la Atrofia muscular (disminución del volumen de los músculos), la Debilidad muscular (disminución de fuerza), las Fasciculaciones (contracciones involuntarias de algunas fibras musculares, dando la apariencia de “latidos”), los Calambres (contracciones dolorosas e involuntarias de un músculo, de breve duración, ocasionando un acortamiento transitorio del mismo), la Espasticidad (aumento del tono muscular, lo cual es percibido como “rígido o duro”), los Trastornos deglutorios (dificultad para tragar los alimentos y los líquidos), la Disartria (dificultad en el habla), el Aumento de la salivación, los Signos Pseudobulbares (llanto y risa inmotivada) y la Insuficiencia respiratoria (dificultad para respirar).
Dependiendo del músculo inicialmente afectado, será donde aparecerá primero la sintomatología.
Los pacientes no tienen trastornos de la sensibilidad como tampoco alteraciones intelectuales.
CAUSAS DE LA ELA:
Aproximadamente de 5 a un 10 % de los pacientes que padecen ELA tienen antecedentes familiares de la enfermedad y corresponden a formas Familiares o Hereditarias y el 90 a 95% restante representan formas Esporádicas (No Hereditarias) de la enfermedad.
Dentro de las formas hereditarias un 20% presentan una alteración (mutación) en el gen (contenido en un cromosoma) que posee la información (codifica) para la producción de una proteína (enzima) llamada Superoxidodismutasa 1 (SOD1). Esta proteína tiene una función dentro de las neuronas motoras, de modo que cuando falta, la neurona muere, siendo esta la causa de la enfermedad. En el 80% restante de las formas Hereditarias aún no se conoce la proteína faltante.
En las formas Esporádicas (no hereditarias) la causa que produce la enfermedad aún no se conoce, es decir que aún no se sabe por qué las neuronas motoras comienzan a degenerar, pero hubo y hay en la actualidad muchos estudios en curso que intentan descubrir la o las causas que provocan la ELA.
Recientemente el profesor Ian Wilmut (quien clonó a la oveja Dolly) en Edimburgo y el profesor Chris Shaw en Londres, solicitaron autorización para clonar embriones humanos para ser usados en la investigación de las causas de las enfermedades de las neuronas motoras, con el objetivo de saber que ocurre en los estadíos tempranos de la enfermedad para poder generar nuevas estrategias de tratamiento.
Aunque no se conoce la causa, se conocen algunos mecanismos que conducen a la degeneración (muerte) de las neuronas motoras en la ELA, uno de ellos y el más relevante hasta la fecha involucra al Glutamato. El Glutamato es una sustancia química (neurotransmisor) que utilizan la neuronas para comunicase entre sí a través de un mecanismo llamado sinapsis. Por alguna razón, aún no bien establecida, en la ELA las neuronas motoras se tornan muy vulnerables al Glutamato, por lo que esta sustancia las daña hasta matarlas.
La posibilidad de identificar y conocer mejor las causas y mecanismos involucrados en el desarrollo de esta enfermedad podría proporcionarnos mejores herramientas para un tratamiento más eficaz, dirigido no solo a controlar los mecanismos sino a combatir la causa o a desarrollar medidas preventivas.
DIAGNÓSTICO EN LA ELA:
El diagnóstico de esta enfermedad es fundamentalmente clínico apoyado por la Electromiografía. Es decir que se basa en el reconocimiento de los síntomas y signos de afectación de la Neurona Motora Superior e inferior y esto solo se logra con un cuidadoso interrogatorio y un prolijo examen neurológico. La electromiografía puede ser de utilidad para detectar signos de compromiso de neurona motora inferior en músculos aún no afectados clínicamente (permitiéndonos conocer la diseminación de la enfermedad).
No existe ningún marcador biológico, es decir, ningún elemento que pueda ser medido o detectado en la sangre ni en ningún otro fluido corporal que permita hacer el diagnóstico de ELA.
Los estudios por imágenes como la Resonancia magnética nuclear o la Tomografía computada tampoco pondrán de manifiesto alteraciones específicas para esta enfermedad, solo nos permiten excluir otras enfermedades que pudieran manifestarse con síntomas de compromiso de neurona motora como en el caso de lesiones medulares, o intracraneales y no siempre es necesaria su realización, reservándoselos solo para cuando existe dudas en el diagnóstico.
Actualmente existen criterios clínicos y electromiográficos para el diagnóstico de la ELA, los cuales fueron establecidos por un grupo de expertos en enfermedades neuromusculares reunidos por la Federación Mundial de Neurología en El Escorial, España, en 1990. Estos criterios son revisados cada 4 años y se modifican de acuerdo a los nuevos conocimientos y avances aportados por la investigación.
Las últimas modificaciones se realizaron en el año 1998 en una reunión similar en Airlie (USA).
En algunas ocasiones el diagnóstico puede resultar dificultoso debido a que el inicio de la enfermedad algunos pacientes manifiestan solo signos de compromiso de la neurona motora superior o inferior pero no combinan ambos elementos como es requerido por los criterios para el diagnóstico de ELA, en estos casos deben realizarse otros exámenes para excluir otras enfermedades.
TRATAMIENTO EN ELA:
No existe en la actualidad un tratamiento que permita curar esta enfermedad. Pero debemos destacar “que una enfermedad no sea curable no quiere decir que no sea tratable”. Si pensamos un poco podremos ver que en la medicina la mayoría de las enfermedades son tratables pero no curables, por ejemplo un paciente con hipertensión arterial necesitará tomar diariamente una medicación para controlar su tensión arterial y si deja el tratamiento lo que mas probablemente ocurra es que nuevamente presente hipertensión arterial, es decir que este paciente es tratado pero no curado. Sí, a diferencia de otras enfermedades, en la ELA aún no se ha logrado un tratamiento etiológico, es decir un tratamiento de la causa, que permita detener la evolución de la enfermedad, actualmente se emplean fármacos que tratan de controlar mecanismos involucrados en la producción de la enfermedad sin lograr aún, a pesar de los múltiples esfuerzos realizados, detener el avance la enfermedad. A este tipo de tratamiento que actúa sobre estos mecanismos lo llamamos Específico, para diferenciarlo del tratamiento Sintomático el cual es dirigido a mejorar los síntomas del paciente y con ello su calidad de vida. Existen muchos medicamentos y procedimientos que pueden aliviar los síntomas que ocasiona la enfermedad como los calambres, el exceso de salivación, la Espasticidad, los problemas deglutorios, respiratorios, etc.
El único fármaco que ha sido aprobado para el tratamiento específico de la ELA, en Argentina y en el resto del mundo es el Riluzole, el cual actúa bloqueando la acción del Glutamato disminuyendo la progresión de la enfermedad, es decir que el Riluzole no cura pero logra retardar la enfermedad. Su efectividad ha sido demostrada mediante estudios correctamente diseñados y controlados.
Muchas otras drogas para el tratamiento de la ELA se encuentran actualmente en desarrollo tales como:
- Neurodex: es una combinación de dos drogas (Dextrometorfan y Sulfato de quinina). Se está estudiando su eficacia para controlar los síntomas Pseudobulbares (llanto y risa inmotivada, explosiva) en pacientes con ELA. Estudios previos ya han demostrado que el Neurodex disminuye estos episodios.
- Celecoxib: es un inhibidor de una enzima llamada ciclooxigenasa-2 (COX-2), que ya ha sido aprobado para el tratamiento de la artritis reumatoidea. La COX-2 interviene en la producción de una sustancia llamada Prostaglandina y esta sustancia estimularía a un grupo de células llamadas Astrocitos, los cuales tienen una función de sostén de las neuronas, para aumentar la liberación de Glutamato, el cual como ya hemos mencionado tiene un efecto tóxico para la neuronas motoras, y además esta enzima induciría la formación de radicales libres, los que también resultan dañinos para las neuronas. Se están desarrollando ensayos terapéuticos para tratar de comprobar si esta droga tiene un efecto neuroprotector, disminuyendo el rango de progresión de la enfermedad.
- Tamoxifeno: es una molécula antiestrogénica, utilizada actualmente para el tratamiento del cáncer de mama. El interés en esta droga como tratamiento para la ELA sobrevino cuando se observó que pacientes con ELA y cáncer de mama, tratadas con Tamoxifeno mostraba una forma mas leve de ELA. Un posible mecanismo por el cual podría provocar este efecto es por la inhibición de una enzima involucrada en producción de Glutamato. Actualmente este estudio se encuentra abierto, incluyendo pacientes para ser tratados con esta droga.
- IGF-1(Factor de crecimiento similar a la insulina): es una sustancia que colabora con el crecimiento de las neuronas. Actualmente se encuentra en estudio y la recibe un numeroso grupo de pacientes en forma subcutánea. El objetivo es determinar si enlentece la progresión de la debilidad en pacientes con ELA. Aún no existen resultados concluyentes.
- Creatina: es una sustancia que mejora la producción de energía en el músculo y estudios en animales han demostrado un efecto neuroprotector. Varios estudios con Creatina en humanos administrada por vía oral se encuentran actualmente en curso.
- Minociclina: muchas células de nuestro cuerpo cuando envejecen o para ser recambiadas son eliminadas por un mecanismo de muerte celular programada llamado apoptosis. La apoptosis es una cascada de eventos que ocurren en las células en los que intervienen enzimas llamadas caspasas, entre otras. La Minociclina es un antibiótico que inhibe a estas enzimas caspasas interviniendo en los mecanismos de apoptosis con lo que se podría retrasar la muerte Neuronal. Ya se ha evidenciado un efecto positivo en animales de laboratorios en los que se le provocó artificialmente una enfermedad de neurona motora. Su estudio en humanos con ELA ha comenzado recientemente.
- Indinavir: es una droga antiviral utilizada actualmente para el tratamiento del HIV. Su mecanismo de acción aún no está muy claro pero parece intervenir en los mecanismos de Apoptosis (muerte celular programada). En pacientes con HIV que desarrollan un síndrome similar a la ELA, este responde al tratamiento con Indinavir. Actualmente se intenta determinar si esta medicación actuaría enlenteciendo la progresión de la ELA en general.
- Antioxidantes: hay evidencias de que en la ELA existe un aumento en la producción de sustancias con acción oxidante que son tóxicas para las neuronas. Por tal motivo se diseñaron estudios donde se evaluó el efecto de sustancias antioxidantes como la vitamina E y C, sin demostrar hasta la fecha un claro beneficio en su empleo.
- Buspirona: es una medicación utilizada comúnmente para estados de ansiedad. Existen evidencias de que los Factores neurotróficos (sustancias que colaboran con el crecimiento y mantenimiento de las neuronas) pueden ser beneficiosos en la ELA. En cuanto al mecanismo de acción de la Buspirona, parece estimular la actividad de Factores neurotróficos endógenos (del propio cuerpo) como el Factor de crecimiento nervioso y el Factor de crecimiento derivado del cerebro. Ya ha demostrado en animales de laboratorio con enfermedad de neurona motora mejorar la función respiratoria. Actualmente está siendo probada en pacientes con ELA.
- Neotrofin: Aún está siendo probado en animales con enfermedades degenerativas como la ELA. Actúa regulando la producción de factores de crecimiento neurotróficos, los cuales son esenciales para el crecimiento, maduración y sobrevida de las células nerviosas. Cuando se encuentran en concentraciones apropiadas, estos factores protegen a las células nerviosas contra el daño producido por enfermedades degenerativas, pero no serían producidos espontáneamente por el sistema nervioso en cantidades suficientes como para ser efectivo en estas condiciones. No hay aún evidencias sobre su efectividad en la ELA.
- NAALADasa: es una enzima que convierte a una sustancia producida en el cerebro en glutamato el cual, como se mencionara anteriormente, tiene un efecto tóxico sobre las neuronas. Se piensa que inhibiendo a esta enzima se reducirían los niveles de glutamato en las enfermedades neurodegenerativas. Aún está siendo probado en cultivos celulares de laboratorio.
Otras posibilidades terapéuticas en desarrollo:
Se encuentran en desarrollo varios estudios en animales y en humanos con Células Madres (Stem cells). Las células madres son células que tienen la capacidad de multiplicarse y transformarse en múltiples tipos de células especializadas como en células de la sangre, hueso, músculo, sistema nervioso.
Se está investigando la posibilidad de colocar células madres en el sistema nervioso e inducirlas a diferenciarse en aquellas células nerviosas dañadas, como en neuronas motoras en el caso de la ELA.
Estas células se pueden obtener de embriones de muy pocos días llamados Blastocistos, de personas adultas (existen células madres en el hígado, medula ósea, piel, corazón, riñón y sistema nervioso) y de sangre de cordón umbilical.
El descubrimiento de células madres capaces de transformarse en neuronas ha abierto una nueva puerta en la reparación cerebral a través del transplante de células madres (embrionarias o adultas) o estimulación de células madres endógenas, es decir residentes en el cerebro adulto, las que serían inducidas a diferenciarse en el tipo neuronal necesario de acuerdo a la patología existente.
Aún los resultados son controvertidos y se requiere de mas investigación para determinar si estas fuentes de células madres pueden ser útiles para la terapéutica en ELA ya que las neuronas motoras tienen un gran desafío para formar conexiones con su blanco (músculo), dado que una vez colocadas en su sitio (cerebro, medula espinal) deben generar prolongaciones (axones) que deben recorrer una gran distancia para llegar hasta los músculos, además el mecanismo de muerte celular en la ELA es poco claro y no se sabe si las células madres transplantadas pueden ser resistentes a la misma fuente de daño que causa la muerte de las neuronas motoras.
Fuente:
http://www.calmo.org.ar
Tema: Algunas consideraciones y actualizaciones sobre el tratamiento de la ELA
GENERALIDADES:
Para realizar un movimiento, como mover un brazo, caminar, tragar o respirar, entre otros, el cerebro debe enviar una orden hacia los músculos involucrados con esa actividad. Para ello cuenta con un grupo de células nerviosas ubicadas en el cerebro, llamadas Neuronas Motoras Superiores, las que constituyen el primer nivel en el circuito motor, por lo que también se las denomina Neuronas Motoras Primarias. Estas Neuronas motoras primarias se conectan y envían señales, a través de un mecanismo denominado Sinapsis, a otras neuronas ubicadas en la medula espinal, la cual se encuentra dentro de la columna vertebral, a las que llamamos Neuronas Motoras Inferiores o Neuronas motoras secundarias. Estas últimas Neuronas establecen contacto con los músculos a través de largas prolongaciones que se reúnen formando los nervios periféricos. Es decir que la orden se origina en el cerebro, viaja por la médula y los nervios periféricos para luego arribar a los músculos que ejecutarán la orden original.
TIPOS DE ENFERMEDAD DE NEURONA MOTORA:
Las enfermedades de la Neurona Motora pueden afectar exclusivamente a las neuronas Motoras Superiores (ubicadas en el cerebro), como es el caso de la Esclerosis lateral primaria (ELP), o selectivamente a las Neuronas Motoras Inferiores (ubicadas en la medula espinal), como por ejemplo las Atrofias espinales, o pueden afectar ambos grupos de neuronas, Superiores e Inferiores, como en el caso de la Esclerosis Lateral Amiotrófica (ELA)
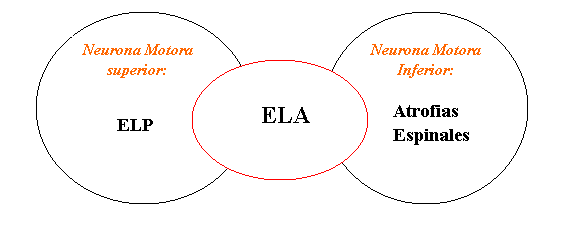 Existen otros tipos de neuronas en el cerebro y la medula espinal, como las que movilizan los ojos, las que transportan la sensibilidad o las que procesan la memoria, las que no son afectadas en este grupo de enfermedades.
Existen otros tipos de neuronas en el cerebro y la medula espinal, como las que movilizan los ojos, las que transportan la sensibilidad o las que procesan la memoria, las que no son afectadas en este grupo de enfermedades.SINTOMAS EN LA ELA:
Los síntomas van apareciendo gradualmente a medida que se enferman las neuronas motoras, entre ellos podemos mencionar la Atrofia muscular (disminución del volumen de los músculos), la Debilidad muscular (disminución de fuerza), las Fasciculaciones (contracciones involuntarias de algunas fibras musculares, dando la apariencia de “latidos”), los Calambres (contracciones dolorosas e involuntarias de un músculo, de breve duración, ocasionando un acortamiento transitorio del mismo), la Espasticidad (aumento del tono muscular, lo cual es percibido como “rígido o duro”), los Trastornos deglutorios (dificultad para tragar los alimentos y los líquidos), la Disartria (dificultad en el habla), el Aumento de la salivación, los Signos Pseudobulbares (llanto y risa inmotivada) y la Insuficiencia respiratoria (dificultad para respirar).
Dependiendo del músculo inicialmente afectado, será donde aparecerá primero la sintomatología.
Los pacientes no tienen trastornos de la sensibilidad como tampoco alteraciones intelectuales.
CAUSAS DE LA ELA:
Aproximadamente de 5 a un 10 % de los pacientes que padecen ELA tienen antecedentes familiares de la enfermedad y corresponden a formas Familiares o Hereditarias y el 90 a 95% restante representan formas Esporádicas (No Hereditarias) de la enfermedad.
Dentro de las formas hereditarias un 20% presentan una alteración (mutación) en el gen (contenido en un cromosoma) que posee la información (codifica) para la producción de una proteína (enzima) llamada Superoxidodismutasa 1 (SOD1). Esta proteína tiene una función dentro de las neuronas motoras, de modo que cuando falta, la neurona muere, siendo esta la causa de la enfermedad. En el 80% restante de las formas Hereditarias aún no se conoce la proteína faltante.
En las formas Esporádicas (no hereditarias) la causa que produce la enfermedad aún no se conoce, es decir que aún no se sabe por qué las neuronas motoras comienzan a degenerar, pero hubo y hay en la actualidad muchos estudios en curso que intentan descubrir la o las causas que provocan la ELA.
Recientemente el profesor Ian Wilmut (quien clonó a la oveja Dolly) en Edimburgo y el profesor Chris Shaw en Londres, solicitaron autorización para clonar embriones humanos para ser usados en la investigación de las causas de las enfermedades de las neuronas motoras, con el objetivo de saber que ocurre en los estadíos tempranos de la enfermedad para poder generar nuevas estrategias de tratamiento.
Aunque no se conoce la causa, se conocen algunos mecanismos que conducen a la degeneración (muerte) de las neuronas motoras en la ELA, uno de ellos y el más relevante hasta la fecha involucra al Glutamato. El Glutamato es una sustancia química (neurotransmisor) que utilizan la neuronas para comunicase entre sí a través de un mecanismo llamado sinapsis. Por alguna razón, aún no bien establecida, en la ELA las neuronas motoras se tornan muy vulnerables al Glutamato, por lo que esta sustancia las daña hasta matarlas.
La posibilidad de identificar y conocer mejor las causas y mecanismos involucrados en el desarrollo de esta enfermedad podría proporcionarnos mejores herramientas para un tratamiento más eficaz, dirigido no solo a controlar los mecanismos sino a combatir la causa o a desarrollar medidas preventivas.
DIAGNÓSTICO EN LA ELA:
El diagnóstico de esta enfermedad es fundamentalmente clínico apoyado por la Electromiografía. Es decir que se basa en el reconocimiento de los síntomas y signos de afectación de la Neurona Motora Superior e inferior y esto solo se logra con un cuidadoso interrogatorio y un prolijo examen neurológico. La electromiografía puede ser de utilidad para detectar signos de compromiso de neurona motora inferior en músculos aún no afectados clínicamente (permitiéndonos conocer la diseminación de la enfermedad).
No existe ningún marcador biológico, es decir, ningún elemento que pueda ser medido o detectado en la sangre ni en ningún otro fluido corporal que permita hacer el diagnóstico de ELA.
Los estudios por imágenes como la Resonancia magnética nuclear o la Tomografía computada tampoco pondrán de manifiesto alteraciones específicas para esta enfermedad, solo nos permiten excluir otras enfermedades que pudieran manifestarse con síntomas de compromiso de neurona motora como en el caso de lesiones medulares, o intracraneales y no siempre es necesaria su realización, reservándoselos solo para cuando existe dudas en el diagnóstico.
Actualmente existen criterios clínicos y electromiográficos para el diagnóstico de la ELA, los cuales fueron establecidos por un grupo de expertos en enfermedades neuromusculares reunidos por la Federación Mundial de Neurología en El Escorial, España, en 1990. Estos criterios son revisados cada 4 años y se modifican de acuerdo a los nuevos conocimientos y avances aportados por la investigación.
Las últimas modificaciones se realizaron en el año 1998 en una reunión similar en Airlie (USA).
En algunas ocasiones el diagnóstico puede resultar dificultoso debido a que el inicio de la enfermedad algunos pacientes manifiestan solo signos de compromiso de la neurona motora superior o inferior pero no combinan ambos elementos como es requerido por los criterios para el diagnóstico de ELA, en estos casos deben realizarse otros exámenes para excluir otras enfermedades.
TRATAMIENTO EN ELA:
No existe en la actualidad un tratamiento que permita curar esta enfermedad. Pero debemos destacar “que una enfermedad no sea curable no quiere decir que no sea tratable”. Si pensamos un poco podremos ver que en la medicina la mayoría de las enfermedades son tratables pero no curables, por ejemplo un paciente con hipertensión arterial necesitará tomar diariamente una medicación para controlar su tensión arterial y si deja el tratamiento lo que mas probablemente ocurra es que nuevamente presente hipertensión arterial, es decir que este paciente es tratado pero no curado. Sí, a diferencia de otras enfermedades, en la ELA aún no se ha logrado un tratamiento etiológico, es decir un tratamiento de la causa, que permita detener la evolución de la enfermedad, actualmente se emplean fármacos que tratan de controlar mecanismos involucrados en la producción de la enfermedad sin lograr aún, a pesar de los múltiples esfuerzos realizados, detener el avance la enfermedad. A este tipo de tratamiento que actúa sobre estos mecanismos lo llamamos Específico, para diferenciarlo del tratamiento Sintomático el cual es dirigido a mejorar los síntomas del paciente y con ello su calidad de vida. Existen muchos medicamentos y procedimientos que pueden aliviar los síntomas que ocasiona la enfermedad como los calambres, el exceso de salivación, la Espasticidad, los problemas deglutorios, respiratorios, etc.
El único fármaco que ha sido aprobado para el tratamiento específico de la ELA, en Argentina y en el resto del mundo es el Riluzole, el cual actúa bloqueando la acción del Glutamato disminuyendo la progresión de la enfermedad, es decir que el Riluzole no cura pero logra retardar la enfermedad. Su efectividad ha sido demostrada mediante estudios correctamente diseñados y controlados.
Muchas otras drogas para el tratamiento de la ELA se encuentran actualmente en desarrollo tales como:
- Neurodex: es una combinación de dos drogas (Dextrometorfan y Sulfato de quinina). Se está estudiando su eficacia para controlar los síntomas Pseudobulbares (llanto y risa inmotivada, explosiva) en pacientes con ELA. Estudios previos ya han demostrado que el Neurodex disminuye estos episodios.
- Celecoxib: es un inhibidor de una enzima llamada ciclooxigenasa-2 (COX-2), que ya ha sido aprobado para el tratamiento de la artritis reumatoidea. La COX-2 interviene en la producción de una sustancia llamada Prostaglandina y esta sustancia estimularía a un grupo de células llamadas Astrocitos, los cuales tienen una función de sostén de las neuronas, para aumentar la liberación de Glutamato, el cual como ya hemos mencionado tiene un efecto tóxico para la neuronas motoras, y además esta enzima induciría la formación de radicales libres, los que también resultan dañinos para las neuronas. Se están desarrollando ensayos terapéuticos para tratar de comprobar si esta droga tiene un efecto neuroprotector, disminuyendo el rango de progresión de la enfermedad.
- Tamoxifeno: es una molécula antiestrogénica, utilizada actualmente para el tratamiento del cáncer de mama. El interés en esta droga como tratamiento para la ELA sobrevino cuando se observó que pacientes con ELA y cáncer de mama, tratadas con Tamoxifeno mostraba una forma mas leve de ELA. Un posible mecanismo por el cual podría provocar este efecto es por la inhibición de una enzima involucrada en producción de Glutamato. Actualmente este estudio se encuentra abierto, incluyendo pacientes para ser tratados con esta droga.
- IGF-1(Factor de crecimiento similar a la insulina): es una sustancia que colabora con el crecimiento de las neuronas. Actualmente se encuentra en estudio y la recibe un numeroso grupo de pacientes en forma subcutánea. El objetivo es determinar si enlentece la progresión de la debilidad en pacientes con ELA. Aún no existen resultados concluyentes.
- Creatina: es una sustancia que mejora la producción de energía en el músculo y estudios en animales han demostrado un efecto neuroprotector. Varios estudios con Creatina en humanos administrada por vía oral se encuentran actualmente en curso.
- Minociclina: muchas células de nuestro cuerpo cuando envejecen o para ser recambiadas son eliminadas por un mecanismo de muerte celular programada llamado apoptosis. La apoptosis es una cascada de eventos que ocurren en las células en los que intervienen enzimas llamadas caspasas, entre otras. La Minociclina es un antibiótico que inhibe a estas enzimas caspasas interviniendo en los mecanismos de apoptosis con lo que se podría retrasar la muerte Neuronal. Ya se ha evidenciado un efecto positivo en animales de laboratorios en los que se le provocó artificialmente una enfermedad de neurona motora. Su estudio en humanos con ELA ha comenzado recientemente.
- Indinavir: es una droga antiviral utilizada actualmente para el tratamiento del HIV. Su mecanismo de acción aún no está muy claro pero parece intervenir en los mecanismos de Apoptosis (muerte celular programada). En pacientes con HIV que desarrollan un síndrome similar a la ELA, este responde al tratamiento con Indinavir. Actualmente se intenta determinar si esta medicación actuaría enlenteciendo la progresión de la ELA en general.
- Antioxidantes: hay evidencias de que en la ELA existe un aumento en la producción de sustancias con acción oxidante que son tóxicas para las neuronas. Por tal motivo se diseñaron estudios donde se evaluó el efecto de sustancias antioxidantes como la vitamina E y C, sin demostrar hasta la fecha un claro beneficio en su empleo.
- Buspirona: es una medicación utilizada comúnmente para estados de ansiedad. Existen evidencias de que los Factores neurotróficos (sustancias que colaboran con el crecimiento y mantenimiento de las neuronas) pueden ser beneficiosos en la ELA. En cuanto al mecanismo de acción de la Buspirona, parece estimular la actividad de Factores neurotróficos endógenos (del propio cuerpo) como el Factor de crecimiento nervioso y el Factor de crecimiento derivado del cerebro. Ya ha demostrado en animales de laboratorio con enfermedad de neurona motora mejorar la función respiratoria. Actualmente está siendo probada en pacientes con ELA.
- Neotrofin: Aún está siendo probado en animales con enfermedades degenerativas como la ELA. Actúa regulando la producción de factores de crecimiento neurotróficos, los cuales son esenciales para el crecimiento, maduración y sobrevida de las células nerviosas. Cuando se encuentran en concentraciones apropiadas, estos factores protegen a las células nerviosas contra el daño producido por enfermedades degenerativas, pero no serían producidos espontáneamente por el sistema nervioso en cantidades suficientes como para ser efectivo en estas condiciones. No hay aún evidencias sobre su efectividad en la ELA.
- NAALADasa: es una enzima que convierte a una sustancia producida en el cerebro en glutamato el cual, como se mencionara anteriormente, tiene un efecto tóxico sobre las neuronas. Se piensa que inhibiendo a esta enzima se reducirían los niveles de glutamato en las enfermedades neurodegenerativas. Aún está siendo probado en cultivos celulares de laboratorio.
Otras posibilidades terapéuticas en desarrollo:
Se encuentran en desarrollo varios estudios en animales y en humanos con Células Madres (Stem cells). Las células madres son células que tienen la capacidad de multiplicarse y transformarse en múltiples tipos de células especializadas como en células de la sangre, hueso, músculo, sistema nervioso.
Se está investigando la posibilidad de colocar células madres en el sistema nervioso e inducirlas a diferenciarse en aquellas células nerviosas dañadas, como en neuronas motoras en el caso de la ELA.
Estas células se pueden obtener de embriones de muy pocos días llamados Blastocistos, de personas adultas (existen células madres en el hígado, medula ósea, piel, corazón, riñón y sistema nervioso) y de sangre de cordón umbilical.
El descubrimiento de células madres capaces de transformarse en neuronas ha abierto una nueva puerta en la reparación cerebral a través del transplante de células madres (embrionarias o adultas) o estimulación de células madres endógenas, es decir residentes en el cerebro adulto, las que serían inducidas a diferenciarse en el tipo neuronal necesario de acuerdo a la patología existente.
Aún los resultados son controvertidos y se requiere de mas investigación para determinar si estas fuentes de células madres pueden ser útiles para la terapéutica en ELA ya que las neuronas motoras tienen un gran desafío para formar conexiones con su blanco (músculo), dado que una vez colocadas en su sitio (cerebro, medula espinal) deben generar prolongaciones (axones) que deben recorrer una gran distancia para llegar hasta los músculos, además el mecanismo de muerte celular en la ELA es poco claro y no se sabe si las células madres transplantadas pueden ser resistentes a la misma fuente de daño que causa la muerte de las neuronas motoras.
Fuente:
http://www.calmo.org.ar
lunes, mayo 29, 2006
La Fundación Favaloro tendrá un nuevo centro de neurociencias
MARTES 23 de Mayo de 2006 - Ciencia / Salud
LANACION.com.
Hoy se presenta en sociedad
La Fundación Favaloro tendrá un nuevo centro de neurociencias
Reúne a un grupo de destacados especialistas; estudiará la relación corazón-cerebro
Para Eduardo Raimondi, vicepresidente y director general de la Fundación Favaloro, era una asignatura pendiente.
Para Facundo Manes, joven neurocientífico que acepta el desafío de conducir a un grupo de destacados especialistas, el cumplimiento de un sueño: ofrecer tratamientos de excelencia a un gran número de pacientes de distinta condición social, como los de obras sociales o los privados subvencionados que en la actualidad no tienen medios para acceder a neurología, psiquiatría y neurocirugía de primer nivel.
Los caminos se cruzaron y hoy se presenta en sociedad un ambicioso proyecto asistencial y científico: el nuevo Instituto de Neurociencias de la Fundación Favaloro, que contará con departamentos de neurología, psiquiatría, neurocirugía y neuropsicología, además de un Centro de Stroke (accidente cerebrovascular) que abarcará desde el tratamiento de la emergencia hasta la prevención secundaria y la rehabilitación.
"Estamos en un muy buen momento en lo que hace a la docencia, la investigacion y la asistencia de máximo nivel -afirma Raimondi-. Y si hasta ahora nos conocían básicamente como un centro de alta tecnología en cardiología y trasplante, de aquí en más el desafío es que nos asocien también con el cerebro."
Manes, por su parte, afirma que se siente honrado de formar parte del proyecto de uno de los argentinos más importantes del siglo XX, como fue René Favaloro. "Mi padre estuvo internado aquí cuando yo era residente -recuerda-. Aquí se atiende a personas de todas las clases sociales, y siempre sentí el desafío de ofrecer opciones de excelencia para todos."
Para comprender la importancia que se asigna al emprendimiento, baste con tener en cuenta que aunque ofrece atención en numerosas especialidades y recibe 12.000 consultas mensuales, hasta hoy la Fundación tenía un solo instituto, el de Cardiología y Cirugía Cardiovascular (Icicc). Es más, los planes para los próximos cinco años contemplan incluso la posibilidad de dotarlo de un edificio propio.
Ejes de investigación
Precisamente, uno de los ejes sobre el que trabajará el equipo del nuevo centro es la relación corazón-cerebro. En ese sentido, algunas de las líneas de investigación se centrarán, por ejemplo, en las relaciones que existen entre enfermedad cardiovascular y trastornos psiquiátricos, entre enfermedad vascular y demencia, entre hipertensión arterial y deterioro cognitivo, y entre estatinas, colesterol y enfermedad de Alzheimer.
Para conducir las distintas áreas de tratamiento e investigación, Manes reunió a un "seleccionado" de la neurología, con especialistas formados en las mejores escuelas locales de la disciplina, como son Fleni, el Hospital Ramos Mejía, el Hospital Francés y el Hospital de Clínicas.
En la conducción del Departamento de Neurología estará el doctor Alberto Dubrovsky, vicepresidente de la Asociación Argentina de Neurología y presidente del próximo Congreso Argentino de Neurología.
El Departamento de Psiquiatría estará a cargo del doctor Marcelo Cetkovich-Bakmas; el de Neurocirugía será dirigido por el doctor Anselmo Rodríguez Loffredo; el de Neuropsicología y Fonoaudiología, por la licenciada Teresa Torralva; el de Emergencias Neurológicas, por el doctor Francisco Klein, y el Centro de Stroke, por los doctores Luciano Sposato y Klein.
Los doctores Manes y Alicia Lischinky dirigirán el Centro de Alzheimer, Deterioro Cognitivo Vascular y Neuropsiquiatría.
"Es un proyecto que nos motiva y todos tenemos un gran entusiasmo -confiesa Manes-. Queremos desarrollar un polo multidisciplinario de trabajo clínico e investigación de referencia internacional, que permita acercarse a la comprensión de los trastornos neurológicos y encontrar estrategias eficaces de tratamiento. Personalmente, dirigí un grupo en Fleni, creé y dirijo el Instituto de Neurología Cognitiva (Ineco), estoy en un buen momento y quería hacer algo por el país. Es una oportunidad que no se iba a dar en dos décadas. Tenía que tomar esta responsabilidad."
Por Nora Bär
De la Redacción de LA NACION
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=808212
Copyright S. A. LA NACION 2006. Todos los derechos reservados.
LANACION.com.
Hoy se presenta en sociedad
La Fundación Favaloro tendrá un nuevo centro de neurociencias
Reúne a un grupo de destacados especialistas; estudiará la relación corazón-cerebro
Para Eduardo Raimondi, vicepresidente y director general de la Fundación Favaloro, era una asignatura pendiente.
Para Facundo Manes, joven neurocientífico que acepta el desafío de conducir a un grupo de destacados especialistas, el cumplimiento de un sueño: ofrecer tratamientos de excelencia a un gran número de pacientes de distinta condición social, como los de obras sociales o los privados subvencionados que en la actualidad no tienen medios para acceder a neurología, psiquiatría y neurocirugía de primer nivel.
Los caminos se cruzaron y hoy se presenta en sociedad un ambicioso proyecto asistencial y científico: el nuevo Instituto de Neurociencias de la Fundación Favaloro, que contará con departamentos de neurología, psiquiatría, neurocirugía y neuropsicología, además de un Centro de Stroke (accidente cerebrovascular) que abarcará desde el tratamiento de la emergencia hasta la prevención secundaria y la rehabilitación.
"Estamos en un muy buen momento en lo que hace a la docencia, la investigacion y la asistencia de máximo nivel -afirma Raimondi-. Y si hasta ahora nos conocían básicamente como un centro de alta tecnología en cardiología y trasplante, de aquí en más el desafío es que nos asocien también con el cerebro."
Manes, por su parte, afirma que se siente honrado de formar parte del proyecto de uno de los argentinos más importantes del siglo XX, como fue René Favaloro. "Mi padre estuvo internado aquí cuando yo era residente -recuerda-. Aquí se atiende a personas de todas las clases sociales, y siempre sentí el desafío de ofrecer opciones de excelencia para todos."
Para comprender la importancia que se asigna al emprendimiento, baste con tener en cuenta que aunque ofrece atención en numerosas especialidades y recibe 12.000 consultas mensuales, hasta hoy la Fundación tenía un solo instituto, el de Cardiología y Cirugía Cardiovascular (Icicc). Es más, los planes para los próximos cinco años contemplan incluso la posibilidad de dotarlo de un edificio propio.
Ejes de investigación
Precisamente, uno de los ejes sobre el que trabajará el equipo del nuevo centro es la relación corazón-cerebro. En ese sentido, algunas de las líneas de investigación se centrarán, por ejemplo, en las relaciones que existen entre enfermedad cardiovascular y trastornos psiquiátricos, entre enfermedad vascular y demencia, entre hipertensión arterial y deterioro cognitivo, y entre estatinas, colesterol y enfermedad de Alzheimer.
Para conducir las distintas áreas de tratamiento e investigación, Manes reunió a un "seleccionado" de la neurología, con especialistas formados en las mejores escuelas locales de la disciplina, como son Fleni, el Hospital Ramos Mejía, el Hospital Francés y el Hospital de Clínicas.
En la conducción del Departamento de Neurología estará el doctor Alberto Dubrovsky, vicepresidente de la Asociación Argentina de Neurología y presidente del próximo Congreso Argentino de Neurología.
El Departamento de Psiquiatría estará a cargo del doctor Marcelo Cetkovich-Bakmas; el de Neurocirugía será dirigido por el doctor Anselmo Rodríguez Loffredo; el de Neuropsicología y Fonoaudiología, por la licenciada Teresa Torralva; el de Emergencias Neurológicas, por el doctor Francisco Klein, y el Centro de Stroke, por los doctores Luciano Sposato y Klein.
Los doctores Manes y Alicia Lischinky dirigirán el Centro de Alzheimer, Deterioro Cognitivo Vascular y Neuropsiquiatría.
"Es un proyecto que nos motiva y todos tenemos un gran entusiasmo -confiesa Manes-. Queremos desarrollar un polo multidisciplinario de trabajo clínico e investigación de referencia internacional, que permita acercarse a la comprensión de los trastornos neurológicos y encontrar estrategias eficaces de tratamiento. Personalmente, dirigí un grupo en Fleni, creé y dirijo el Instituto de Neurología Cognitiva (Ineco), estoy en un buen momento y quería hacer algo por el país. Es una oportunidad que no se iba a dar en dos décadas. Tenía que tomar esta responsabilidad."
Por Nora Bär
De la Redacción de LA NACION
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=808212
Copyright S. A. LA NACION 2006. Todos los derechos reservados.
Es bueno para los músculos el ácido láctico
Ciencia/Salud
Martes 23 de Mayo de 2006
Deportistas
Es bueno para los músculos el ácido láctico
NUEVA YORK (The New York Times).- Cualquiera que al menos haya pensado en hacer gimnasia ha escuchado advertencias acerca del ácido láctico. Se acumula en los músculos. Es lo que quema los músculos. Su acumulación es lo que hace que los músculos se cansen y se dañen.
Los entrenadores les dicen a los atletas que tienen que aprender a trabajar justo por debajo del "umbral láctico", cuando la sustancia comienza a acumularse. Algunos hasta se hacen tests sanguíneos para conocer sus umbrales lácticos personales.
Pero resulta que eso está todo mal. El ácido láctico es en realidad un combustible, no un producto de desecho. Los músculos lo hacen deliberadamente, a partir de la glucosa, y lo queman para obtener energía. La razón de que los atletas pueden esforzarse tan fuertemente y durante tanto tiempo es que la práctica hace que sus músculos absorban más eficientemente el ácido láctico. "Es uno de los errores clásicos de la historia de la ciencia", afirma el doctor George Brooks, profesor del departamento de biología integrativa de la Universidad de California en Berkeley.
Los orígenes de este malentendido surgen del estudio de un premio Nobel, Otto Meyerhof, que a principios del siglo XX cortó un sapo por la mitad y puso la parte inferior en una jarra. Los músculos no tenían circulación ni fuente de oxígeno. Cuando Myerhoff los examinó, descubrió que estaban bañados en ácido láctico. Había nacido una teoría: la falta de oxígeno conduce al ácido láctico, que conduce a la fatiga.
Hoy se sabe que las células musculares convierten la glucosa en glicógeno o ácido láctico. Este es absorbido y utilizado como combustible por las mitocondrias, las fábricas de energía de las células. Las mitocondrias incluso tienen una proteína especial para transportarlo a su interior, según descubrió Brooks.
El entrenamiento intenso hace una gran diferencia porque puede duplicar la masa de las mitocondrias y hacer que éstas quemen más ácido láctico y sus músculos puedan trabajar más duramente y durante más tiempo.
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=808214
LA NACION | 23.05.2006 | Página 14 | Ciencia/Salud
Copyright 2006 SA LA NACION | Todos los derechos reservados
Martes 23 de Mayo de 2006
Deportistas
Es bueno para los músculos el ácido láctico
NUEVA YORK (The New York Times).- Cualquiera que al menos haya pensado en hacer gimnasia ha escuchado advertencias acerca del ácido láctico. Se acumula en los músculos. Es lo que quema los músculos. Su acumulación es lo que hace que los músculos se cansen y se dañen.
Los entrenadores les dicen a los atletas que tienen que aprender a trabajar justo por debajo del "umbral láctico", cuando la sustancia comienza a acumularse. Algunos hasta se hacen tests sanguíneos para conocer sus umbrales lácticos personales.
Pero resulta que eso está todo mal. El ácido láctico es en realidad un combustible, no un producto de desecho. Los músculos lo hacen deliberadamente, a partir de la glucosa, y lo queman para obtener energía. La razón de que los atletas pueden esforzarse tan fuertemente y durante tanto tiempo es que la práctica hace que sus músculos absorban más eficientemente el ácido láctico. "Es uno de los errores clásicos de la historia de la ciencia", afirma el doctor George Brooks, profesor del departamento de biología integrativa de la Universidad de California en Berkeley.
Los orígenes de este malentendido surgen del estudio de un premio Nobel, Otto Meyerhof, que a principios del siglo XX cortó un sapo por la mitad y puso la parte inferior en una jarra. Los músculos no tenían circulación ni fuente de oxígeno. Cuando Myerhoff los examinó, descubrió que estaban bañados en ácido láctico. Había nacido una teoría: la falta de oxígeno conduce al ácido láctico, que conduce a la fatiga.
Hoy se sabe que las células musculares convierten la glucosa en glicógeno o ácido láctico. Este es absorbido y utilizado como combustible por las mitocondrias, las fábricas de energía de las células. Las mitocondrias incluso tienen una proteína especial para transportarlo a su interior, según descubrió Brooks.
El entrenamiento intenso hace una gran diferencia porque puede duplicar la masa de las mitocondrias y hacer que éstas quemen más ácido láctico y sus músculos puedan trabajar más duramente y durante más tiempo.
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=808214
LA NACION | 23.05.2006 | Página 14 | Ciencia/Salud
Copyright 2006 SA LA NACION | Todos los derechos reservados
viernes, mayo 26, 2006
Consultas Neumonológicas: 19/09/2003
C.A.L.Mo.
Notas sobre la Conferencia del Dr. Eduardo De Vito (médico neumonólogo) realizada en C.A.L.Mo., el día 19 de septiembre de 2003.
El Doctor desempeña sus tareas en el Laboratorio Pulmonar, y en la Evaluación de pacientes neurológicos con compromiso de los músculos respiratorios, en el Instituto de Investigaciones Médicas Alfredo Lanari, organismo dependiente de la Universidad de Buenos Aires. Atiende, casi exclusivamente a pacientes neurológicos con distrofias musculares y E.L.A. Es además, Director Médico de la “Clínica Del Parque”, centro de rehabilitación respiratoria.
Tema: consultas Neumonológicas
Información general de orientación para comprender algunos problemas que produce la enfermedad.
PRESTAR ATENCIÓN A:
* DEGLUCIÓN
* SALIVACIÓN
* CAMBIOS EN LA VOZ
* TOS
* DIFICULTAD PARA RESPIRAR O DISEÑA (ahogo, falta de aire)
LA SALIVACIÓN
* Es muy molesta y genera angustia al paciente. Limita la capacidad para comer porque junto con la comida el paciente se atraganta, come menos, baja de peso y se debilita. Hay que tratar de romper el círculo de descenso de peso-desnutrición-debilidad.
* El inicio de estos problemas es variable y depende de cada paciente. Algunos inician la enfermedad con estos síntomas y otros los tienen a los dos, tres o cinco años de diagnosticada la enfermedad.
* Existen medidas para disminuir la salivación.
o El antidepresivo amitriptilina (recetado por el médico neurólogo), tiene el efecto colateral de producir sequedad en la boca, y se usa con buenos resultados en general.
o Polvo de PAPAYA: (venta libre) se utiliza debajo de la lengua.
o PROPANTENINA: (con receta médica)
o Ciertas GOTAS OFTÁLMICAS: (con receta médica) que contienen atropínicos se utilizan debajo de la lengua
o IRRADIACIÓN DE LAS PAROTIDAS. En casos muy rebeldes a los otros tratamientos. Producen disminución de la cantidad de saliva de las glándulas parótidas.
* Todas estas medidas terapéuticas, combinadas o separadas, en distintas dosis, sirven para tratar de disminuir la cantidad de saliva. Esto es muy importante porque si la saliva entra a la traquea puede producir complicaciones infecciosas.
LA DEGLUCIÓN
* Está en relación con la saliva. Existe una estrategia de alimentos: sólidos, semi-sólidos y distintas consistencias y espesantes que tienden a que el bolo alimenticio pase más fácil para el esófago y no se introduzca en la tráquea.
LA TOS
* Tiene mucho que ver con la deglución. Es el tema más importante. La tos esta debilitada porque hay debilidad en los músculos abdominales.
* La kinesioterapia respiratoria es necesaria en estos casos.
* Existen varias estrategias para la tos:
o NEBULIZACIONES para fluidificar las secreciones y permitir que salgan las flemas más fácilmente. Las nebulizaciones dependen de la condición del paciente, se pueden repetir varias veces al día. No se recomienda usar equipos ultrasónicos porque no nebulizan bien en estos casos. No debe utilizarse careta para nebulizar porque al respirar por la nariz, el líquido no llega a los pulmones, sino que condensa en la nariz, que actúa como filtro. La nebulización tiene que hacerse por la boca entreabierta. Se recomienda no usar la máscara sino la pipeta de vidrio o de plástico del equipo. La boca tiene que estar entreabierta y no cerrada para que entre aire de afuera para favorecer la nebulización.
o KINESIOTERAPIA RESPIRATORIA en sus dos aspectos: profesional y familiar. En determinados momentos de la enfermedad, cuando hay muchas secreciones es el kinesiólogo el que tiene que acudir en su ayuda. Cuando se va el profesional, quien cuida al paciente tiene que conocer básicamente el tema para continuar con las maniobras. Son muy importantes posiciones determinadas para favorecer la tos y ayudar a toser apretando el abdomen (consultar con el kinesiólogo para esta maniobra). La mejor medicación es la hidratación: tomar no menos de 2 litros de líquidos por día y las nebulizaciones. Los jarabes para la tos no son efectivos para este caso y los antitusivos están contraindicados porque pueden empeorar el cuadro al inhibir la tos, que es un mecanismo reflejo para expulsar las secreciones y proteger la vía aérea.
o SONDA o GASTROSTOMIA son dos opciones para la alimentación cuando ésta se hace muy dificultosa por boca y el paciente tose y se aspira. Así se mantiene los niveles de nutrición y se evita que lo que coma se aspire al pulmón. La experiencia en el mundo indica que los pacientes que no aceptan en el momento oportuno la gastrostomía, se debilitan más rápidamente. La gastrostomía, está claramente comprobado, que mejora la calidad de vida de los pacientes con E.L.A. Consiste en una punción en el abdomen, se coloca una sonda anclada que sale al exterior con un taponcito. Queda enroscado en la piel y se desenrosca para conectarla con la guía para la alimentación por donde pasa el alimento o líquidos. Es una sonda flexible y la intervención puede ser quirúrgica, con anestesia o bien con endoscopía, con anestesia más suave (recomendable), del tipo de la utilizada para endoscopías altas por úlceras. Es una operación que requiere quizá 24 hs de internación .
o La decisión de colocar una sonda o gastrostomía, tiene que estar consensuada con el paciente. Es voluntad del paciente, quien decide si desea o no efectuarla.
o El monitoreo del PESO es muy importante porque, cuando se conjugan la salivación, la mala deglución, el atragantarse y la pérdida de peso, se puede estar necesitando una gastrostomía. Poner una gastrostomía, no significa que no se pueda comer por boca nunca más. Es muy importante saber esto. Lo que se debe lograr es que pase por la gastrostomía, el agua y las calorías necesarias, mientras que por boca, se ingieren aquellos alimentos que son más fáciles y que producen placer.
CONSULTAS VARIAS
* EL FRÍO
o A veces el paciente tiene mala regulación térmica porque hay menos músculos que protejan, al igual que la grasa. Se debe constatar que no haya otra enfermedad que produzca la sensación de frío o que tenga fiebre (cuadro infeccioso) y se lo debe abrigar para darle confort.
* CANSANCIO
o Es normal que el paciente se queje de cansancio ya que la enfermedad produce debilidad muscular y produce cansancio en respuesta a esfuerzos mínimos, como caminar de un cuarto a otro. La idea es adecuar la actividad para no cansarse. Hay que buscar un punto intermedio que es la posibilidad del paciente en términos realistas: no exigirle que camine cuando se fatiga y mantener la movilidad según las posibilidades.
o LA OBESIDAD El sobrepeso empeora las dificultades motoras. Pero el paciente históricamente “gordito”, si pierde peso en forma no controlada perderá grasa y músculo. Hay que lograr una dieta adecuada que le permita bajar la grasa sin perder el músculo.
LAS DIETAS
o No hay ninguna dieta que preserve o aumente la masa muscular. La dieta tiene que estar calculada en base a las calorías estimadas de pérdida de peso y de actividad. Tiene que tener contenido proteico elevado, no exagerado. Los trabajos realizados sobre anabólicos y hormonas anabólicas, en los años 70 y 80 no mostraron resultados positivos.
* LOS EJERCICIOS
o Si la enfermedad está estable, se puede hacer un plan de ejercicios de rehabilitación no contra peso. Si no hay estabilidad, no se puede realizar ejercicios que dañen al músculo. El plan de ejercicios es mantener las funciones, la movilidad y lo que el paciente tiene al día. El movimiento de los ojos no suele ser problemático, sólo cuando la enfermedad está muy avanzada pueden comenzar los problemas oculares. En caso de afectarse, no hay ejercicios para esto. Se pueden realizar ejercicios sin problemas, porque los músculos de los ojos no trabajan contra carga o sea que no se dañan con el ejercicio excesivo.
* TUBO DE OXIGENO
o Eventualmente se puede necesitar un tubo de oxígeno en la casa, para las nebulizaciones, para cuando se atraganta o cuando siente falta de aire. Disminuye la ansiedad del paciente.
* DIFICULTAD RESPIRATORIA
o Asegurarse que no haya mucosidad que la produzca o que se haya atragantado.
o USO DE OXIGENO en puntuales circunstancias.
o USO DE BI-PAP (ventilación no invasiva) es una máscara hermética que permite insuflar aire al paciente, en forma cíclica. El efecto puede durar varias horas después. Está indicado por el médico neumonólogo en base a estudios funcionales como la espirometría, en base a la oxigenación de la sangre y la fuerza muscular respiratoria, que pueda tener el paciente. Muchos pacientes lo usan algunas horas durante el día o durante la noche, dependiendo de cada caso y la progresión de la enfermedad. Es un tema que debe ser hablado con el paciente, si desea o no usar este sistema. La tendencia mundial es usar la BI-PAP en detrimento de la TRAQUEOSTOMIA (utilizada hace unos 20 años atrás).
Fuente:
http://www.calmo.org.ar
Notas sobre la Conferencia del Dr. Eduardo De Vito (médico neumonólogo) realizada en C.A.L.Mo., el día 19 de septiembre de 2003.
El Doctor desempeña sus tareas en el Laboratorio Pulmonar, y en la Evaluación de pacientes neurológicos con compromiso de los músculos respiratorios, en el Instituto de Investigaciones Médicas Alfredo Lanari, organismo dependiente de la Universidad de Buenos Aires. Atiende, casi exclusivamente a pacientes neurológicos con distrofias musculares y E.L.A. Es además, Director Médico de la “Clínica Del Parque”, centro de rehabilitación respiratoria.
Tema: consultas Neumonológicas
Información general de orientación para comprender algunos problemas que produce la enfermedad.
PRESTAR ATENCIÓN A:
* DEGLUCIÓN
* SALIVACIÓN
* CAMBIOS EN LA VOZ
* TOS
* DIFICULTAD PARA RESPIRAR O DISEÑA (ahogo, falta de aire)
LA SALIVACIÓN
* Es muy molesta y genera angustia al paciente. Limita la capacidad para comer porque junto con la comida el paciente se atraganta, come menos, baja de peso y se debilita. Hay que tratar de romper el círculo de descenso de peso-desnutrición-debilidad.
* El inicio de estos problemas es variable y depende de cada paciente. Algunos inician la enfermedad con estos síntomas y otros los tienen a los dos, tres o cinco años de diagnosticada la enfermedad.
* Existen medidas para disminuir la salivación.
o El antidepresivo amitriptilina (recetado por el médico neurólogo), tiene el efecto colateral de producir sequedad en la boca, y se usa con buenos resultados en general.
o Polvo de PAPAYA: (venta libre) se utiliza debajo de la lengua.
o PROPANTENINA: (con receta médica)
o Ciertas GOTAS OFTÁLMICAS: (con receta médica) que contienen atropínicos se utilizan debajo de la lengua
o IRRADIACIÓN DE LAS PAROTIDAS. En casos muy rebeldes a los otros tratamientos. Producen disminución de la cantidad de saliva de las glándulas parótidas.
* Todas estas medidas terapéuticas, combinadas o separadas, en distintas dosis, sirven para tratar de disminuir la cantidad de saliva. Esto es muy importante porque si la saliva entra a la traquea puede producir complicaciones infecciosas.
LA DEGLUCIÓN
* Está en relación con la saliva. Existe una estrategia de alimentos: sólidos, semi-sólidos y distintas consistencias y espesantes que tienden a que el bolo alimenticio pase más fácil para el esófago y no se introduzca en la tráquea.
LA TOS
* Tiene mucho que ver con la deglución. Es el tema más importante. La tos esta debilitada porque hay debilidad en los músculos abdominales.
* La kinesioterapia respiratoria es necesaria en estos casos.
* Existen varias estrategias para la tos:
o NEBULIZACIONES para fluidificar las secreciones y permitir que salgan las flemas más fácilmente. Las nebulizaciones dependen de la condición del paciente, se pueden repetir varias veces al día. No se recomienda usar equipos ultrasónicos porque no nebulizan bien en estos casos. No debe utilizarse careta para nebulizar porque al respirar por la nariz, el líquido no llega a los pulmones, sino que condensa en la nariz, que actúa como filtro. La nebulización tiene que hacerse por la boca entreabierta. Se recomienda no usar la máscara sino la pipeta de vidrio o de plástico del equipo. La boca tiene que estar entreabierta y no cerrada para que entre aire de afuera para favorecer la nebulización.
o KINESIOTERAPIA RESPIRATORIA en sus dos aspectos: profesional y familiar. En determinados momentos de la enfermedad, cuando hay muchas secreciones es el kinesiólogo el que tiene que acudir en su ayuda. Cuando se va el profesional, quien cuida al paciente tiene que conocer básicamente el tema para continuar con las maniobras. Son muy importantes posiciones determinadas para favorecer la tos y ayudar a toser apretando el abdomen (consultar con el kinesiólogo para esta maniobra). La mejor medicación es la hidratación: tomar no menos de 2 litros de líquidos por día y las nebulizaciones. Los jarabes para la tos no son efectivos para este caso y los antitusivos están contraindicados porque pueden empeorar el cuadro al inhibir la tos, que es un mecanismo reflejo para expulsar las secreciones y proteger la vía aérea.
o SONDA o GASTROSTOMIA son dos opciones para la alimentación cuando ésta se hace muy dificultosa por boca y el paciente tose y se aspira. Así se mantiene los niveles de nutrición y se evita que lo que coma se aspire al pulmón. La experiencia en el mundo indica que los pacientes que no aceptan en el momento oportuno la gastrostomía, se debilitan más rápidamente. La gastrostomía, está claramente comprobado, que mejora la calidad de vida de los pacientes con E.L.A. Consiste en una punción en el abdomen, se coloca una sonda anclada que sale al exterior con un taponcito. Queda enroscado en la piel y se desenrosca para conectarla con la guía para la alimentación por donde pasa el alimento o líquidos. Es una sonda flexible y la intervención puede ser quirúrgica, con anestesia o bien con endoscopía, con anestesia más suave (recomendable), del tipo de la utilizada para endoscopías altas por úlceras. Es una operación que requiere quizá 24 hs de internación .
o La decisión de colocar una sonda o gastrostomía, tiene que estar consensuada con el paciente. Es voluntad del paciente, quien decide si desea o no efectuarla.
o El monitoreo del PESO es muy importante porque, cuando se conjugan la salivación, la mala deglución, el atragantarse y la pérdida de peso, se puede estar necesitando una gastrostomía. Poner una gastrostomía, no significa que no se pueda comer por boca nunca más. Es muy importante saber esto. Lo que se debe lograr es que pase por la gastrostomía, el agua y las calorías necesarias, mientras que por boca, se ingieren aquellos alimentos que son más fáciles y que producen placer.
CONSULTAS VARIAS
* EL FRÍO
o A veces el paciente tiene mala regulación térmica porque hay menos músculos que protejan, al igual que la grasa. Se debe constatar que no haya otra enfermedad que produzca la sensación de frío o que tenga fiebre (cuadro infeccioso) y se lo debe abrigar para darle confort.
* CANSANCIO
o Es normal que el paciente se queje de cansancio ya que la enfermedad produce debilidad muscular y produce cansancio en respuesta a esfuerzos mínimos, como caminar de un cuarto a otro. La idea es adecuar la actividad para no cansarse. Hay que buscar un punto intermedio que es la posibilidad del paciente en términos realistas: no exigirle que camine cuando se fatiga y mantener la movilidad según las posibilidades.
o LA OBESIDAD El sobrepeso empeora las dificultades motoras. Pero el paciente históricamente “gordito”, si pierde peso en forma no controlada perderá grasa y músculo. Hay que lograr una dieta adecuada que le permita bajar la grasa sin perder el músculo.
LAS DIETAS
o No hay ninguna dieta que preserve o aumente la masa muscular. La dieta tiene que estar calculada en base a las calorías estimadas de pérdida de peso y de actividad. Tiene que tener contenido proteico elevado, no exagerado. Los trabajos realizados sobre anabólicos y hormonas anabólicas, en los años 70 y 80 no mostraron resultados positivos.
* LOS EJERCICIOS
o Si la enfermedad está estable, se puede hacer un plan de ejercicios de rehabilitación no contra peso. Si no hay estabilidad, no se puede realizar ejercicios que dañen al músculo. El plan de ejercicios es mantener las funciones, la movilidad y lo que el paciente tiene al día. El movimiento de los ojos no suele ser problemático, sólo cuando la enfermedad está muy avanzada pueden comenzar los problemas oculares. En caso de afectarse, no hay ejercicios para esto. Se pueden realizar ejercicios sin problemas, porque los músculos de los ojos no trabajan contra carga o sea que no se dañan con el ejercicio excesivo.
* TUBO DE OXIGENO
o Eventualmente se puede necesitar un tubo de oxígeno en la casa, para las nebulizaciones, para cuando se atraganta o cuando siente falta de aire. Disminuye la ansiedad del paciente.
* DIFICULTAD RESPIRATORIA
o Asegurarse que no haya mucosidad que la produzca o que se haya atragantado.
o USO DE OXIGENO en puntuales circunstancias.
o USO DE BI-PAP (ventilación no invasiva) es una máscara hermética que permite insuflar aire al paciente, en forma cíclica. El efecto puede durar varias horas después. Está indicado por el médico neumonólogo en base a estudios funcionales como la espirometría, en base a la oxigenación de la sangre y la fuerza muscular respiratoria, que pueda tener el paciente. Muchos pacientes lo usan algunas horas durante el día o durante la noche, dependiendo de cada caso y la progresión de la enfermedad. Es un tema que debe ser hablado con el paciente, si desea o no usar este sistema. La tendencia mundial es usar la BI-PAP en detrimento de la TRAQUEOSTOMIA (utilizada hace unos 20 años atrás).
Fuente:
http://www.calmo.org.ar
miércoles, mayo 24, 2006
Ensayo clínico con células madre de la médula ósea en Elche, España
Dado que, al parecer, todavía no han seleccionado a los 10 pacientes que participarán en el ensayo clínico con células madre de la médula ósea, en Elche, escribí a la Fundación Diógenes para preguntar cuales son los requisitos para entrar a formar parte del ensayo. Les copio la respuesta:
Quisiera saber como te encuentras, como va siendo la evolución de la enfermedad. Como sabes sólo son 10 afectados. envía por correo postal tu historial clínico y desde la fundación enviaremos todos los historiales de los que dispongamos a los equipos que van a realizar el ensayo. Nuestra filosofía es que todo afectado que lo desee tiene derecho a que su historial sea evaluado.
Dirección:
Fundación Diógenes para la Investigación de la ELA.
C/ Bernabé del Campo Latorre, 26
03202 ELCHE (Alicante)
Tl 96.542.48.66
Quedamos a tu disposición
Elena Contreras.
info@fundacionela.com
http://www.fundacionela.com
Gentileza de Pilar G
Quisiera saber como te encuentras, como va siendo la evolución de la enfermedad. Como sabes sólo son 10 afectados. envía por correo postal tu historial clínico y desde la fundación enviaremos todos los historiales de los que dispongamos a los equipos que van a realizar el ensayo. Nuestra filosofía es que todo afectado que lo desee tiene derecho a que su historial sea evaluado.
Dirección:
Fundación Diógenes para la Investigación de la ELA.
C/ Bernabé del Campo Latorre, 26
03202 ELCHE (Alicante)
Tl 96.542.48.66
Quedamos a tu disposición
Elena Contreras.
info@fundacionela.com
http://www.fundacionela.com
Gentileza de Pilar G
XLIII Congreso Argentino de Neurología
Programa Preliminar
MAR DEL PLATA 2006
15 al 18 de noviembre de 2006
Carta de Bienvenida
Autoridades
Cursos Post-Congreso
Actividades Científicas
Costos de inscripción
Informes e Inscripción
Bienvenidos!
Estimados colegas y demás profesionales de la salud:
La SNA ya está trabajando intensamente en la organización del XLIII Congreso Argentino de Neurología.
El éxito de las convocatorias previas nos ha animado a abordar esta nueva edición
con singular entusiasmo.
En los últimos 4 años, acompañando el crecimiento de nuestra Sociedad, hemos aumentado en un 100% el número de participantes a nuestro evento más importante
lo que nos coloca frente a un nuevo desafío.
Trabajaremos para que este congreso sea también “para todos”: neurólogos clínicos, colegas de otras especialidades, electrofisiólogos, investigadores básicos y auxiliares
de la medicina.
Aspiramos a organizar un congreso tanto para miembros de nuestra Sociedad como
de otras sociedades afines, interesados en el estudio de las diversas áreas de las neurociencias.
El congreso es también el ámbito apropiado para encontrarse con amigos y hacer nuevos, es el lugar propicio para conectarse, planear, discutir proyectos, transmitir y nutrirse de experiencias, escuchar y ser escuchados.
La reunión será provechosa no solamente para los que ya venimos transitando el ejercicio de la medicina por muchos años sino que va dirigido muy especialmente para aquellos
que siendo más jóvenes, están empezando a trazar un camino que será a su vez transitado por los que les sigan.
Los “Cursos del Congreso” de singular éxito en la edición pasada renovarán la apuesta este año.
Mar del Plata y la sede elegida (Sheraton Hotel) ofrecen el marco ideal para conjugar
todo esto con las posibilidades turísticas excepcionales que todos conocemos.
En nombre de la SNA y su Comisión Directiva, de los Comités Científico y Organizador
del XLIII Congreso Argentino de Neurología y en el mío propio los invito a asistir y participar, durante los días 15 al XX 18 Noviembre de 2006 de sus sesiones científicas y actos sociales. También los invito a que juntos podamos ilusionarnos y comprometernos desde este momento en su planificación y desarrollo, ya que aspiramos a que este congreso sea de Ustedes.
La participación de todos enriquecerá su contenido.
Los Esperamos!
Prof. Dr. Alberto L Dubrovsky
Presidente XLIII Congreso Argentino de Neurología
Autoridades
Presidente
Dr. Alberto Dubrovsky
Vicepresidente
Dr. Mario Melcon
Secretarios
Dr. Sergio Gonorazky
Dr. Ricardo Allegri
Tesorero
Dr. Roberto Rey
Presidente Comité Organizador
Dr. Horacio E. Gori
Miembros
Dr. Juan José Cirio
Dr. Diego Dato
Dra. Laura Díaz
Dr. Daniel Gestro
Dr. Daniel Muñoz
Dr. Luis Quintas
Presidente Comité Científico
Dr. Jorge Correale
Miembros
Dr. Sebastián Ameriso
Dr. Lucas Bonamico
Dra. María Laura Garau
Dra. Silvia García
Dr. Oscar Gershanik
Dr. Francisco Gualtieri
Dra. Silvia Kochen
Dra. Lidia López
Dr. Ricardo Reisin
Dr. Carlos Rugilo
Dr. Luciano Sposato
CURSOS POST-CONGRESO: sábado 18 de noviembre
3 h. de duración con puntaje para la recertificación en la especialidad.
NO ARANCELADOS - INSCRIPCION EN SECRETARIA
1- Manejo agudo y prevención del Stroke 2006
Directores: Dr. Sebastián Ameriso - Dr. Luciano Sposato
2- Importancia del diagnóstico diferencial de las Demencias en la evolución, pronóstico y tratamiento
Directora: Dra. María Laura Garau
3- Cefaleas: consensos en diagnóstico y terapéutica
Director: Dr. Lucas Bonamico
4- Mareos y vértigo
Director: Dr. Francisco Gualtieri
5- Estrategias terapéuticas en Movimientos Anormales: Distonías-Tics y Coreas
Directores: Dra. Silvia García - Dr. Oscar Gershanik
6- ¿Cómo estamos seguros que el paciente tiene Epilepsia?
Directora: Dra. Silvia Kochen
7- Emergencias en Neurología
Director: Dr. Jorge Correale
8- Patología Neuromuscular
Director: Dr. Ricardo Reisin
Actividades Científicas
Conferencias Plenarias, Mesas Redondas, Simposios, Talleres, Sesiones de Posters y Plataformas.
Temas:
• Cefaleas
• Demencias
• Enfermedades Cerebrovasculares
• Neuro-rehabilitación
• Dolor neuropático
• Epilepsia
• Esclerosis Múltiple
• Movimientos Anormales
• Neuro-oftalmología y Neuro-otología
• Alteraciones de la marcha
• Patología regional (Tuberculosis, Chagas, Cisticercosis, y Enfermedad de Hansen)
• Medicina del sueño
• Neuro-oncología
• Cuidados paliativos
• Neuro-genética
• Avances terapéuticos en Demencia, Dolor, Enfermedad de Parkinson y Epilepsia
Costos de inscripción
Inscripción General $ 150.-
Socios de la S.N.A $ 100.-
Residentes / Concurrentes $ 50.-
Técnicos y estudiantes $ 20.-
Inscripción diaria $ 30.-
Material de Cursos del Congreso
Socio S.N.A. inscripto al curso sin cargo
Socios S.N.A no inscripto $ 10.-
Material para no socio $ 20.-
Informes e inscripción
Lic. Romina Gori
Tel: (5411) 4837-9687
E-mail: snaeventos@arnet.com.ar
Sociedad Neurológica Argentina
Thames 2127-C1425FIC- Buenos Aires, Argentina
Tel/Fax: (5411) 4773-5850 o (5411) 4899-0582
E-mail: snaeventos@arnet.com.ar
Website: www.sna.org.ar
La Sociedad Neurológica Argentina no ha designado, en esta ocasión, Agencia de Turismo Oficial para dicho evento, quedando librada a su propia elección la contratación de la Hotelería.
Le sugerimos consultar la siguiente página web: www.mardelplata.com
MAR DEL PLATA 2006
15 al 18 de noviembre de 2006
Carta de Bienvenida
Autoridades
Cursos Post-Congreso
Actividades Científicas
Costos de inscripción
Informes e Inscripción
Bienvenidos!
Estimados colegas y demás profesionales de la salud:
La SNA ya está trabajando intensamente en la organización del XLIII Congreso Argentino de Neurología.
El éxito de las convocatorias previas nos ha animado a abordar esta nueva edición
con singular entusiasmo.
En los últimos 4 años, acompañando el crecimiento de nuestra Sociedad, hemos aumentado en un 100% el número de participantes a nuestro evento más importante
lo que nos coloca frente a un nuevo desafío.
Trabajaremos para que este congreso sea también “para todos”: neurólogos clínicos, colegas de otras especialidades, electrofisiólogos, investigadores básicos y auxiliares
de la medicina.
Aspiramos a organizar un congreso tanto para miembros de nuestra Sociedad como
de otras sociedades afines, interesados en el estudio de las diversas áreas de las neurociencias.
El congreso es también el ámbito apropiado para encontrarse con amigos y hacer nuevos, es el lugar propicio para conectarse, planear, discutir proyectos, transmitir y nutrirse de experiencias, escuchar y ser escuchados.
La reunión será provechosa no solamente para los que ya venimos transitando el ejercicio de la medicina por muchos años sino que va dirigido muy especialmente para aquellos
que siendo más jóvenes, están empezando a trazar un camino que será a su vez transitado por los que les sigan.
Los “Cursos del Congreso” de singular éxito en la edición pasada renovarán la apuesta este año.
Mar del Plata y la sede elegida (Sheraton Hotel) ofrecen el marco ideal para conjugar
todo esto con las posibilidades turísticas excepcionales que todos conocemos.
En nombre de la SNA y su Comisión Directiva, de los Comités Científico y Organizador
del XLIII Congreso Argentino de Neurología y en el mío propio los invito a asistir y participar, durante los días 15 al XX 18 Noviembre de 2006 de sus sesiones científicas y actos sociales. También los invito a que juntos podamos ilusionarnos y comprometernos desde este momento en su planificación y desarrollo, ya que aspiramos a que este congreso sea de Ustedes.
La participación de todos enriquecerá su contenido.
Los Esperamos!
Prof. Dr. Alberto L Dubrovsky
Presidente XLIII Congreso Argentino de Neurología
Autoridades
Presidente
Dr. Alberto Dubrovsky
Vicepresidente
Dr. Mario Melcon
Secretarios
Dr. Sergio Gonorazky
Dr. Ricardo Allegri
Tesorero
Dr. Roberto Rey
Presidente Comité Organizador
Dr. Horacio E. Gori
Miembros
Dr. Juan José Cirio
Dr. Diego Dato
Dra. Laura Díaz
Dr. Daniel Gestro
Dr. Daniel Muñoz
Dr. Luis Quintas
Presidente Comité Científico
Dr. Jorge Correale
Miembros
Dr. Sebastián Ameriso
Dr. Lucas Bonamico
Dra. María Laura Garau
Dra. Silvia García
Dr. Oscar Gershanik
Dr. Francisco Gualtieri
Dra. Silvia Kochen
Dra. Lidia López
Dr. Ricardo Reisin
Dr. Carlos Rugilo
Dr. Luciano Sposato
CURSOS POST-CONGRESO: sábado 18 de noviembre
3 h. de duración con puntaje para la recertificación en la especialidad.
NO ARANCELADOS - INSCRIPCION EN SECRETARIA
1- Manejo agudo y prevención del Stroke 2006
Directores: Dr. Sebastián Ameriso - Dr. Luciano Sposato
2- Importancia del diagnóstico diferencial de las Demencias en la evolución, pronóstico y tratamiento
Directora: Dra. María Laura Garau
3- Cefaleas: consensos en diagnóstico y terapéutica
Director: Dr. Lucas Bonamico
4- Mareos y vértigo
Director: Dr. Francisco Gualtieri
5- Estrategias terapéuticas en Movimientos Anormales: Distonías-Tics y Coreas
Directores: Dra. Silvia García - Dr. Oscar Gershanik
6- ¿Cómo estamos seguros que el paciente tiene Epilepsia?
Directora: Dra. Silvia Kochen
7- Emergencias en Neurología
Director: Dr. Jorge Correale
8- Patología Neuromuscular
Director: Dr. Ricardo Reisin
Actividades Científicas
Conferencias Plenarias, Mesas Redondas, Simposios, Talleres, Sesiones de Posters y Plataformas.
Temas:
• Cefaleas
• Demencias
• Enfermedades Cerebrovasculares
• Neuro-rehabilitación
• Dolor neuropático
• Epilepsia
• Esclerosis Múltiple
• Movimientos Anormales
• Neuro-oftalmología y Neuro-otología
• Alteraciones de la marcha
• Patología regional (Tuberculosis, Chagas, Cisticercosis, y Enfermedad de Hansen)
• Medicina del sueño
• Neuro-oncología
• Cuidados paliativos
• Neuro-genética
• Avances terapéuticos en Demencia, Dolor, Enfermedad de Parkinson y Epilepsia
Costos de inscripción
Inscripción General $ 150.-
Socios de la S.N.A $ 100.-
Residentes / Concurrentes $ 50.-
Técnicos y estudiantes $ 20.-
Inscripción diaria $ 30.-
Material de Cursos del Congreso
Socio S.N.A. inscripto al curso sin cargo
Socios S.N.A no inscripto $ 10.-
Material para no socio $ 20.-
Informes e inscripción
Lic. Romina Gori
Tel: (5411) 4837-9687
E-mail: snaeventos@arnet.com.ar
Sociedad Neurológica Argentina
Thames 2127-C1425FIC- Buenos Aires, Argentina
Tel/Fax: (5411) 4773-5850 o (5411) 4899-0582
E-mail: snaeventos@arnet.com.ar
Website: www.sna.org.ar
La Sociedad Neurológica Argentina no ha designado, en esta ocasión, Agencia de Turismo Oficial para dicho evento, quedando librada a su propia elección la contratación de la Hotelería.
Le sugerimos consultar la siguiente página web: www.mardelplata.com
martes, mayo 23, 2006
Avicena Anuncia Resultados Del Ensayo De la Fase III ALS
Avicena Anuncia Resultados Del Ensayo De la Fase III ALS
ALS-02 Demuestra Tendencia Positiva De la Mortalidad
Palo Alto, California, 9 de mayo, 2006 /PRNewswire-FirstCall/ -- El Avicena Group, Inc. (Avicena), un revelador de los productos farmacéuticos y terapéuticos de la novela, anuncia hoy los resultados de un ensayo de la fase III de su droga, ALS-02, para el tratamiento de la Esclerosis Lateral Amiotrófica (enfermedad de ALS o de Lou Gehrig). Los datos del estudio no demostraron ninguna diferencia estadística significativa entre ALS-02 (cinco gramos por día) y el placebo con respecto a los puntos finales primarias y secundarias del estudio (varias medidas de fuerza muscular, de fatiga del músculo y de cuentas funcionales). Sin embargo, en un análisis separado cuando los datos de la mortalidad fueron combinados y analizados con datos de la mortalidad de un segundo estudio de ALS-02 conducido previamente por el North East ALS Consortium (NEALS), el análisis demostró una tendencia positiva hacia mortalidad disminuida en los temas que recibían ALS-02. El compuesto también es encontrado para ser seguro y tolerado bien por los pacientes.
"Estamos muy interesados en explorar más lejos esta tendencia hacia mortalidad disminuida," dijo Belinda Tsao-Nivaggioli, Ph.D., ejecutiva de Avicena. "Nos preponemos explorar si estos resultados se pueden esperar para afectar la trayectoria reguladora restante para el compuesto."
En un análisis separado, los investigadores están combinando actualmente datos sobre todas las medidas del resultado del ensayo recientemente terminado de la fase III, conducido por Jeffrey Rosenfeld, M.D., Ph.D., director del Carolinas ALS Center, con datos del ensayo previamente terminado de la fase II de ALS-02, liderado por Jeremy Shefner, de M.D. y conducido por NEALS. Un meta-análisis comprensivo también se está planeando para combinar datos de todos los ensayos formales en los cuales ALS-02 fue utilizado como agente terapéutico experimental en ALS. Sobre la terminación del meta-análisis, Avicena se prepone discutir los resultados, incluyendo la tendencia de la mortalidad, con la United States Food and Drug Administration (FDA) para determinar los pasos siguientes apropiados del desarrollo para ALS-02.
"Creemos que junto con el FDA podremos identificar una trayectoria clara para poner en el mercado ALS-02," indica esperanzada la Dr. Tsao-Nivaggioli. "Es importante observar que un tratamiento actualmente aprobado para los pacientes que sufren de ALS ofrece solamente una limitada ventaja a corto plazo de la supervivencia y no ha demostrado la ventaja en vista de medidas de la fuerza muscular y de la función neurológica."
Además, en los estudios anteriores conducidos en otras condiciones neurodegenerativas tales como enfermedad de Huntington, los datos sugieren que la terapéutica creatina-basada propietaria de Avicena puede ser considerablemente más eficaz en dosis más altas, sin causar un aumento en efectos nocivos. Como tal, el Dr. Allitia DiBernardo y el consorcio de NEALS, con Avicena, están planeando actualmente un estudio de la dosis-escalada de ALS-02, que se está diseñando para determinar la dosificación óptima del tratamiento dentro de la población de paciente de ALS.
El estudio de la fase III de Avicena de ALS-02 fue un multi-center, double-blind, placebo-controlled, seleccionado al azar diseñado para evaluar la seguridad y la eficacia del compuesto en una dosis de cinco gramos por día por nueve meses. El ensayo fue apoyado por el National Institutes of Health's National Center for Complementary and Alternative Medicine. ALS-02 es un terapéutico propietario que incorpora una forma ultra-pura, clínica de creatina. Al compuesto le fue concedida la designación huérfana de la droga por FDA en 2002.
Fuente:
http://www.pharmalive.com/
Traducción:
http://ar.babelfish.yahoo.com/
ALS-02 Demuestra Tendencia Positiva De la Mortalidad
Palo Alto, California, 9 de mayo, 2006 /PRNewswire-FirstCall/ -- El Avicena Group, Inc. (Avicena), un revelador de los productos farmacéuticos y terapéuticos de la novela, anuncia hoy los resultados de un ensayo de la fase III de su droga, ALS-02, para el tratamiento de la Esclerosis Lateral Amiotrófica (enfermedad de ALS o de Lou Gehrig). Los datos del estudio no demostraron ninguna diferencia estadística significativa entre ALS-02 (cinco gramos por día) y el placebo con respecto a los puntos finales primarias y secundarias del estudio (varias medidas de fuerza muscular, de fatiga del músculo y de cuentas funcionales). Sin embargo, en un análisis separado cuando los datos de la mortalidad fueron combinados y analizados con datos de la mortalidad de un segundo estudio de ALS-02 conducido previamente por el North East ALS Consortium (NEALS), el análisis demostró una tendencia positiva hacia mortalidad disminuida en los temas que recibían ALS-02. El compuesto también es encontrado para ser seguro y tolerado bien por los pacientes.
"Estamos muy interesados en explorar más lejos esta tendencia hacia mortalidad disminuida," dijo Belinda Tsao-Nivaggioli, Ph.D., ejecutiva de Avicena. "Nos preponemos explorar si estos resultados se pueden esperar para afectar la trayectoria reguladora restante para el compuesto."
En un análisis separado, los investigadores están combinando actualmente datos sobre todas las medidas del resultado del ensayo recientemente terminado de la fase III, conducido por Jeffrey Rosenfeld, M.D., Ph.D., director del Carolinas ALS Center, con datos del ensayo previamente terminado de la fase II de ALS-02, liderado por Jeremy Shefner, de M.D. y conducido por NEALS. Un meta-análisis comprensivo también se está planeando para combinar datos de todos los ensayos formales en los cuales ALS-02 fue utilizado como agente terapéutico experimental en ALS. Sobre la terminación del meta-análisis, Avicena se prepone discutir los resultados, incluyendo la tendencia de la mortalidad, con la United States Food and Drug Administration (FDA) para determinar los pasos siguientes apropiados del desarrollo para ALS-02.
"Creemos que junto con el FDA podremos identificar una trayectoria clara para poner en el mercado ALS-02," indica esperanzada la Dr. Tsao-Nivaggioli. "Es importante observar que un tratamiento actualmente aprobado para los pacientes que sufren de ALS ofrece solamente una limitada ventaja a corto plazo de la supervivencia y no ha demostrado la ventaja en vista de medidas de la fuerza muscular y de la función neurológica."
Además, en los estudios anteriores conducidos en otras condiciones neurodegenerativas tales como enfermedad de Huntington, los datos sugieren que la terapéutica creatina-basada propietaria de Avicena puede ser considerablemente más eficaz en dosis más altas, sin causar un aumento en efectos nocivos. Como tal, el Dr. Allitia DiBernardo y el consorcio de NEALS, con Avicena, están planeando actualmente un estudio de la dosis-escalada de ALS-02, que se está diseñando para determinar la dosificación óptima del tratamiento dentro de la población de paciente de ALS.
El estudio de la fase III de Avicena de ALS-02 fue un multi-center, double-blind, placebo-controlled, seleccionado al azar diseñado para evaluar la seguridad y la eficacia del compuesto en una dosis de cinco gramos por día por nueve meses. El ensayo fue apoyado por el National Institutes of Health's National Center for Complementary and Alternative Medicine. ALS-02 es un terapéutico propietario que incorpora una forma ultra-pura, clínica de creatina. Al compuesto le fue concedida la designación huérfana de la droga por FDA en 2002.
Fuente:
http://www.pharmalive.com/
Traducción:
http://ar.babelfish.yahoo.com/
lunes, mayo 22, 2006
Proyecto A.L.S. Abre el laboratorio de investigación Privado-Financiado de la célula de vástago, el primero enfocado exclusivamente en ALS
Proyecto A.L.S. Abre el laboratorio de investigación Privado-Financiado de
la célula de vástago, el primero enfocado exclusivamente en ALS y
enfermedades relacionadas
la célula de vástago, el primero enfocado exclusivamente en ALS y
enfermedades relacionadas
El laboratorio del proyecto A.L.S./Jenifer Estess para la investigación
de la célula de vástago se abre hoy en Nueva York.
NUEVA YORK, 15 de mayo /PRNewswire/ El proyecto A.L.S. abre hoy
el primer laboratorio privado-financiado enfocado exclusivamente en
el estudio de las células de vástago para tratar ALS (Esclerosis Lateral
Amiotrófica) y las enfermedades relacionadas a la motoneurona, anunció
Valerie Estess, directora de la investigación para el proyecto A.L.S.
El laboratorio del proyecto A.L.S./Jenifer Estess para la investigación de
la célula de vástago es una empresa a riesgo compartido entre el
proyecto A.L.S. y la universidad de Columbia. Basado en Nueva
York, el proyecto A.L.S. no acepta el financiamiento federal y tiene una
política de "puertas abiertas" que anime a investigadores y a colaboradores
de la célula de vástago del proyecto A.L.S.- de la universidad de Harvard,
de la universidad Johns Hopkins, del instituto Salk, del Memorial Sloan-Kettering
Cancer Center, y de otras instituciones radicadas en Nueva York a que colaboren
con los científicos y los clínicos de la Universidad de Columbia.
"La creación de un laboratorio avanzado en donde las ideas más recientes de la
biología de la célula de vástago se pueden explorar en las neuronas derivadas
humanas del motor de la célula de vástago, sin obstáculo o constreñimiento, es un
logro muy notable," dijo Thomas M. Jessell, Ph.D., investigador médico del instituto
Howard Hughes, profesor de bioquímica y de biofísica molecular y miembro del centro
para la neurobiología y del comportamiento en el centro médico de la
universidad de Columbia en New York City. "La comunidad entera
de ALS, dentro de New York City y nacionalmente, debe al proyecto
A.L.S. una gran gratitud. Su agenda previsora y pro-activa de la investigación está fijando los
estándares para los acercamientos modernos a investigar enfermedades
neurodegenerativas." Christopher E. Henderson, Ph.D., y Hynek
Wichterle, Ph.D., de la universidad de Columbia actuarán como
consejeros científicos mayores al proyecto A.L.S.
El Dr. Jessell será el consejero de la investigación.
Estas citas reconocen la relación de muchos años entre neurólogos renombrados
mundialmente y el proyecto A.L.S de la universidad de
Columbia. En 1999, el proyecto A.L.S. reclutó al laboratorio
del Dr. Jessell para examinar cómo las células de vástago podrían
ayudar a científicos a entender mejor y a tratar ALS. Desde ese
tiempo, lod Drs. Jessell y Wichterle han demostrado
que las células de vástago embrionarias se pueden ordenar para
distinguir en las neuronas funcionales del motor, las mismas células
del nervio destruidas selectivamente en ALS.
El Proyecto A.L.S.de la célula de vástago se abre hoy en Nueva York.
NUEVA YORK, 15 de mayo /PRNewswire/ El proyecto A.L.S. abre hoy
el primer laboratorio privado-financiado enfocado exclusivamente en
el estudio de las células de vástago para tratar ALS (Esclerosis Lateral
Amiotrófica) y las enfermedades relacionadas a la motoneurona, anunció
Valerie Estess, directora de la investigación para el proyecto A.L.S.
El laboratorio del proyecto A.L.S./Jenifer Estess para la investigación de
la célula de vástago es una empresa a riesgo compartido entre el
proyecto A.L.S. y la universidad de Columbia. Basado en Nueva
York, el proyecto A.L.S. no acepta el financiamiento federal y tiene una
política de "puertas abiertas" que anime a investigadores y a colaboradores
de la célula de vástago del proyecto A.L.S.- de la universidad de Harvard,
de la universidad Johns Hopkins, del instituto Salk, del Memorial Sloan-Kettering
Cancer Center, y de otras instituciones radicadas en Nueva York a que colaboren
con los científicos y los clínicos de la Universidad de Columbia.
"La creación de un laboratorio avanzado en donde las ideas más recientes de la
biología de la célula de vástago se pueden explorar en las neuronas derivadas
humanas del motor de la célula de vástago, sin obstáculo o constreñimiento, es un
logro muy notable," dijo Thomas M. Jessell, Ph.D., investigador médico del instituto
Howard Hughes, profesor de bioquímica y de biofísica molecular y miembro del centro
para la neurobiología y del comportamiento en el centro médico de la
universidad de Columbia en New York City. "La comunidad entera
de ALS, dentro de New York City y nacionalmente, debe al proyecto
A.L.S. una gran gratitud. Su agenda previsora y pro-activa de la investigación está fijando los
estándares para los acercamientos modernos a investigar enfermedades
neurodegenerativas." Christopher E. Henderson, Ph.D., y Hynek
Wichterle, Ph.D., de la universidad de Columbia actuarán como
consejeros científicos mayores al proyecto A.L.S.
El Dr. Jessell será el consejero de la investigación.
Estas citas reconocen la relación de muchos años entre neurólogos renombrados
mundialmente y el proyecto A.L.S de la universidad de
Columbia. En 1999, el proyecto A.L.S. reclutó al laboratorio
del Dr. Jessell para examinar cómo las células de vástago podrían
ayudar a científicos a entender mejor y a tratar ALS. Desde ese
tiempo, lod Drs. Jessell y Wichterle han demostrado
que las células de vástago embrionarias se pueden ordenar para
distinguir en las neuronas funcionales del motor, las mismas células
del nervio destruidas selectivamente en ALS.
El laboratorio proporciona un lugar dedicado para la
exploración de estos resultados y para el trabajo hacia tratamientos
y curaciones para las personas con ALS y enfermedades relacionadas a la
motoneurona. El Dr. Jessell continua diciendo, "al establecer el
laboratorio, el proyecto A.L.S. ha pavimentado la manera para una
aceleración dramática en el paso en el cual los avances en biología
básica de la motoneurona se pueden traducir en terapias
clínicas más eficaces para el tratamiento de la Esclerosis
Lateral Amiotrófica y de enfermedades relacionadas."
Inicialmente, las iniciativas de la investigación del proyecto
A.L.S. incluirá:
- El desarrollo de los primeros análisis de celulas basadas en humanos
o de las pruebas de ALS, que proporcionarán la nueva información sobre
la forma humana de la enfermedad. (previamente, los científicos han estudiado los
ratones transgénicos que desarrollan ALS.);
- El uso de las células del vástago derivadas de la motoneurona
para prevenir ALS y a SMA (Atrofia Muscular Espinal);
Posteriormente, el trabajo del proyecto A.L.S. se orientará hacia el
desarrollo de los estudios del trasplante de la célula de vástago. El laboratorio del
proyecto A.L.S./Jenifer Estess para la investigación de la célula de
vástago se denomina asi en memoria de la co-fundadora del proyecto A.L.S.,
Jenifer Estess, que fue diagnosticada con ALS a los treinta y cinco años,
y que murió de la enfermedad en 2003.
ALS es una enfermedad neurodegenerativa uniformemente fatal relacionada con
Alzheimer y Parkinson, y cuyos efectos son parálisis progresiva de casi
todos los músculos voluntarios, incluyendo los que controlan el movimiento, discurso,
y respiración. También conocida como enfermedad de Lou Gehrig, ALS afecta a más de 300.000 americanos. "El Laboratorio Proyecto A.L.S. será un recurso crucial y es extraordinario que el proyecto A.L.S. ha puesto esto en ejecución tan rápidamente. Esto es un buen presagio para el desarrollo acelerado de la terapéutica de la célula de vástago en
ALS", dijo Robert H. Brown, Jr, M.D., D.Phil., profesor de neurología en la escuela médica de Harvard, y un colaborador del proyecto A.L.S.. El proyecto A.L.S. tiene experiencia en
apoyar la investigación prometedora en sus primeros tiempos. En
1998, el proyecto A.L.S. era el primero para unir y para financiar a un
grupo de funcionamiento de biólogos de la célula de vástago, de
clínicos de ALS, de anatomistas, y de científicos básicos para
entender mejor si las células de vástago podrían ser una
herramienta valiosa para abrir los misterios de ALS y de enfermedades
relacionadas. En solamente siete años, el consorcio de la
célula de vástago del proyecto A.L.S. ha alcanzado las brechas
científicas significativas, incluyendo:
- En 2002, Hynek Wichterle (Universidad de Columbia). demostró que las células
de vástago se podrían ordenar para convertirse en motoneuronas
del fide del hueso, las mismas células que se destruyen en ALS y las
enfermedades relacionadas de la neurona del motor (célula, 2002,
agosto 9)
- En 2003, un equipo del proyecto A.L.S.-fconducido
por Doug Kerr y los investigadores de la Universidad Johns Hopkins
demostraron que las ratas con ALS-como la función
significativa recuperada síndrome del motor después de recibir una
infusión de las células de vástago en el líquido espinal
(neurología del diario, el 2003 de de junio 15).
- El colaborador Robert M. Brownstone demostró que las células de vástago
embrionarias derivadas de la motoneurona podrían establecer
conexiones apropiadas con el músculo como objetivo (Diario de la Neurología,
el 8/9/2004; Diario de la Neurología, 22/03/ 2006).
- Los colaboradores incluyendo los Drs. Fred Gage (Instituto Salk), Christopher Kintner (Instituto Salk) y Sally Temple (Universidad Médica de Albany) están caracterizando las poblaciones endógenas de las células de vástago y están anticipando a usar
estas células como herramientas hacia la regeneración y la reparación.
"Proyecto A.L.S. se emociona al abrir el primer
laboratorio de la célula de vástago de su clase, no para las
campanas y los silbidos que vienen junto con una apertura magnífica,
pero porque el proyecto A.L.S. ha demostrado que las células de
vástago ayudarán muy probablemente a entender y tratar una
enfermedad humana fatal", dice Valerie Estess. "El Laboratorio Proyecto
A.L.S. es nuestra última y mas significativa inversión en la investigación de la célula de
vástago. Es el paso siguiente necesario en descubrimiento
científico de avanzada, y, un día, deteniendo las enfermedades del
cerebro."
Sobre El Proyecto A.L.S.
Proyecto A.L.S. es una entidad sin fines de lucro fundada en 1998, por Jenifer Estess,
sus hermanas, y sus amigos, cuando Jenifer fue diagnosticada con ALS
(Esclerosis Lateral Amiotrófica). La mayoría de ingresos de la fundación
son destinados a apoyar investigaciones en genéticas y terapias génicas, células de vástago, la identificación de los caminos de la
enfermedad, y el descubrimiento acelerado de la droga. El sello
de la investigación del proyecto A.L.S. es colaboración. Los
investigadores y los clínicos que antes eran competidores ahora
juegan en el mismo equipo, y trabajan racional, constructiva y
agresivamente hacia las metas de translación más dinámicas, a
saber, los primeros tratamientos eficaces para ALS.
Más información sobre el proyecto A.L.S. se puede encontrar en
http://www.projectals.org
Rachel Martin/Edelman
Oficina: 323.202.1031
Móvil: 323.893.9047
SOURCE Project A.L.S.
Web Site: http://www.projectals.org
Fuente:
http://www.prnewswire.com
Traducción:
http://ar.babelfish.yahoo.com/
viernes, mayo 19, 2006
Una nueva manera de vivir
¿Quién soy?

Mi nombre es Luisa Amelia, venezolana, de 48 años; trabajaba como profesora, me deleite en ello durante 25 hermosos años…; disfrutaba: caminar, trotar, jugar baloncesto, andar en bicicleta, pasear en moto, correr, saltar, nadar, bailar, cantar, conducir, explorar, dar charlas …talleres, asistir a eventos educativos, viajar… Ahora me encuentro limitada para este tipo de actividades.

Pero puedo hacer muchas otras cosas que también disfruto con mucho placer… leer, escribir, cocinar, armar rompecabezas, ir al cine, jugar ajedrez, escuchar música, disfrutar de un buen paseo, investigar sobre temas que me atraen, tomar fotografías, pintar, dibujar, bordar, cuidar mis plantas y sobre todo… disfrutar de mi familia y los amigos.. ELLOS son mi mayor tesoro!!
Mi vida con esta íntima amiga llamada…
E.L.A.
E.L.A.
Hace algún tiempo, mi vida dio un giro inesperado… conocí esta amiga, que ni siquiera sabía que vagaba por el mundo y mucho menos que me elegiría a mí como su compañera…
Mis horas azules se tornaron intensas; pensaba… imaginaba… aceptaba… rechazaba… todo un juego de emociones…
Hasta que me dije:… “si esto es una lotería y el premio gordo te correspondió a ti… tienes que aprender a vivir con ello”…
Inicie mi camino!!!
Mi familia





Esta es mi familia, mis pajaritos como cariñosamente les digo…
Es la familia que he formado…
Desconocíamos todo lo referente a E.L.A.
Una gran nube se posó sobre nuestras cabezas… que hacer???
Comenzamos a investigar, indagar, preguntar... para conocerla mejor...
Ni modo!!! enfrentar… aceptar y vivir!!!
Comprendimos que la cotidianidad del día es lo más importante… que la esperanza brilla en el corazón… y la vida, un hermoso milagro!!
Todo se transformo en aprender a vivir
El mundo es mío!!!

Crecemos, aprendemos, vivimos con cada nueva situación que se nos presenta.
Como yo vea mi mundo con ELA… influirá en ellos y los ayudará a comprenderlo mejor…
La vida tiene hermosos matices, los caminos de Dios son misteriosos, es toda una aventura descubrir sus misterios… crecemos en la corriente de la vida.
Miedos... Temores... Luchas internas???
Por supuesto que los hay, pero no podemos permitir que se apoderen de nuestra paz interior, la aceptación es el mejor camino…
…“Si logramos estar conectados con esa paz interna, sin duda podremos transitar el camino de nuestros destinos acompañados de esa luz, de esa esencia donde ELA no llega”
Nada viene por azar…
Algo debemos aprender de todo esto… la vida no termina con la muerte… y todo ser humano desde que nace, vive muriendo en cada amanecer…
Dios nos eligió por alguna razón… esto nos hace sencillamente especiales… Sí!!! Especiales…
Por qué Especiales???
A pesar de nuestras limitaciones… poseemos fe, esperanza, amor… estamos llenos de vida!!! Tenemos nuestro propio mundo, en el cual libramos batallas diarias con alegría, entusiasmo, perseverancia y una gran fortaleza espiritual… Sin olvidarnos de nuestra capacidad de soñar.
Valoramos cada minuto de nuestra existencia, no somos sumisos ante ELA; ella podrá apoderarse de nuestro cuerpo… pero nuestro corazón, mente y espíritu nos pertenece… la vida es bella y es una hermosa aventura vivirla… Es nuestro reto!!! La vida vale, cuando tenemos el valor de enfrentarla

En todo momento tenemos la oportunidad de aprender algo nuevo, conscientes de que nuestra actitud positiva determina cuán bien aprendemos… La vida en sí, es un aula de clases, que cambia con el ir y venir de las circunstancias. Al ir con la corriente no podemos evitar el aprender y cambiar con cada nueva experiencia; descubriendo en cada una de ellas las maravillas de Dios…




“Si no puedes caminar, usa el bastón, pero sigue siempre hacia delante”
Madre Teresa de Calcuta

Que tengan un radiante día, pleno de alegría!!!!
Reciban un afectuoso abrazo de optimismo desde Venezuela!!!!
Con Amor
Luisa Amelia
cuamashi@yahoo.com
jueves, mayo 18, 2006
Presentación Carolina Filippi
Sr. Carlos Fernández:
Soy Carolina Filippi, tengo ELA diagonsticada con nombre y apellido hace casi dos años
pero sintomas tenia antes.
Vivo en Olavarria, provincia de Bs.As.
Me atiendo en Bs.As. con el Dr. Sica.
Hace dos años que me paso buscando en la web las causa , la medicación y como deben comportarse las prepagas.
Soy una persona mayor, agrimensora, sigo teniendo mi estudio, eso me ayuda a superar este trance.
Me gustaría intercambiar mail con personas que tengan esta enfermedad.
En Olavarrria hay 2 o 3 casos pero hay gente que no le dicen bien como es todo esto.
Yo estoy completamente al tanto.
Voy a un instituto de rehabilitación y a una fonoaudiologa, los dos conocen muy bien el tema.
Tengo afectada la pierna izquierda y en parte la cuerdas vocales. me empezó atrofiándose el pie.
Me interesa intercambiar mail de algunos parendere cosa de esta enfermedad y a otro podré ayudarlos.
Tengo bajada información de ADELA España.
Tambien estoy al tanto sobre certificados de discapacidad, que trae algunas ventajas economicas ya que es una enfermedad muy cara y no todos lo pueden afrontar.
Por ahora te saludo y te agradezco que se esten ocupando de este tema.
Los ayudaré en lo que pueda.
Atentamente. Muchas gracias.
Carolina Filippi
Teléfono 02284-446559
carolinafilippi@yahoo.com.ar
Olavarria
Soy Carolina Filippi, tengo ELA diagonsticada con nombre y apellido hace casi dos años
pero sintomas tenia antes.
Vivo en Olavarria, provincia de Bs.As.
Me atiendo en Bs.As. con el Dr. Sica.
Hace dos años que me paso buscando en la web las causa , la medicación y como deben comportarse las prepagas.
Soy una persona mayor, agrimensora, sigo teniendo mi estudio, eso me ayuda a superar este trance.
Me gustaría intercambiar mail con personas que tengan esta enfermedad.
En Olavarrria hay 2 o 3 casos pero hay gente que no le dicen bien como es todo esto.
Yo estoy completamente al tanto.
Voy a un instituto de rehabilitación y a una fonoaudiologa, los dos conocen muy bien el tema.
Tengo afectada la pierna izquierda y en parte la cuerdas vocales. me empezó atrofiándose el pie.
Me interesa intercambiar mail de algunos parendere cosa de esta enfermedad y a otro podré ayudarlos.
Tengo bajada información de ADELA España.
Tambien estoy al tanto sobre certificados de discapacidad, que trae algunas ventajas economicas ya que es una enfermedad muy cara y no todos lo pueden afrontar.
Por ahora te saludo y te agradezco que se esten ocupando de este tema.
Los ayudaré en lo que pueda.
Atentamente. Muchas gracias.
Carolina Filippi
Teléfono 02284-446559
carolinafilippi@yahoo.com.ar
Olavarria
miércoles, mayo 17, 2006
PRESENTACION EDUARDO BAUERNFEIND
¡¡ HOLA AMIGOS Y AMIGAS !!
Soy Eduardo Bauernfeind, llevo casi 12 meses diagnosticado de una enfermedad que quiero dar a conocer : La Esclerosis Lateral Amiotrófica (ELA).
La ELA es degenerativa, progresiva e irreversible, siendo la más grave del sistema nervioso central. Ataca sólo a las neuronas motoras. Se desconoce su origen y por ahora no tiene ningún tratamiento curativo, no es contagiosa.
La frecuencia es de 2 por cada 100.000 habitantes
Según avanza la enfermedad, se van atrofiando y paralizando de forma lenta los músculos del cuerpo (movimiento de brazos, piernas, control del tronco y cuello) se va perdiendo la capacidad de hablar, tragar y de respirar.
No daña órganos ni afecta la capacidad intelectual ni los sentidos. El conocido científico británico Stephen Hawking también la padece y solo mueve sus ojos.

Tengo 48 años, casado, con dos hijos, un varón de 17 años y una niña de 14 años, vivo en Eldorado, Misiones, Argentina, trabajo como Bioquímico, aunque a partir del 1 de enero del 2006 deje de trabajar porque me es muy penoso caminar, las escaleras son casi insalvables, y la mano izquierda responde cada vez menos.

Los primeros síntomas fueron en abril de 2005 (aunque llevaba algún tiempo antes notándolo) empecé con calambres, cansancio, dolores musculares… notaba que cada día empeoraba más. Llegaron las caídas sin poder controlar las piernas ,¡¡ al suelo !!, tuve muchas caídas , el miedo se apoderó de mí.
El primer Dr. que me dijo: tiene pinta de esclerosis Lateral Amioatrofica fue D. Rito Quintana, Neurólogo de Posadas Misiones, Entre pruebas y Médicos pasó el tiempo; estaba desesperado…cada vez estaba peor y más asustado,
El día del diagnóstico de certeza (principios de junio de 2005), que me lo hizo el Dr. Dubrovsky Alberto , Neurólogo en Buenos Aires,( profesional de gran tacto y calidad humana), fue el peor de mi vida, me sentía hundido, vacío por dentro, inútil, impotente……Mi cabeza daba vueltas siempre a lo mismo: soy una carga, ¿porqué a mí?, ¿porqué me envían éste castigo?, ¿veré crecer a mis hijos?, ¿cuánto me queda?, ¿como voy a poder con esto?
Conducir es cada vez más difícil, los planes, sueños e ilusiones que tienes se esfuman.
Algunos familiares y amigos se olvidan de que existes, pero no importa, tienes amigos/as que te demuestran que lo son, conoces nuevas amigas que te visitan y se preocupan por ti, en Eldorado me lo han demostrado y lo agradezco mucho, porque veo que todavía hay personas de buen corazón que te quieren por como eres, no por tu enfermedad,. GRACIAS POR la AYUDA Y EL CARIÑO QUE ME DAN.
Ahora estoy mejor, me e dado cuenta que debo seguir para adelante disfrutando cada segundo de mi vida al máximo y valorando todo lo que me rodea. Intento ayudar a nuevos afectados, que están asustados, a vivir el presente y no pensar en el futuro, ¡¡ así me siento útil !!.
ESCLEROSIS
LATERAL
AMIOTRÓFICA
CON ESTE FOLLETO QUIERO QUE CONOZCAN ESTA ENFERMEDAD Y COMO HA CAMBIADO MI VIDA; A MI ESPOSA QUE VA SUPERÁNDOLO POCO A POCO Y A MIS HIJOS QUE SON MUY VALIENTES; LOS QUIERO MUCHO.
Me gustaría conocer afectados de ELA en Argentina , si conocen a alguien y quiere conversar: Tel 03751-421466 o 03751-15666922 / e- mail : eduabauer@yahoo.com.ar o eduardo_bauernfeind@hotmail.com
Soy Eduardo Bauernfeind, llevo casi 12 meses diagnosticado de una enfermedad que quiero dar a conocer : La Esclerosis Lateral Amiotrófica (ELA).
La ELA es degenerativa, progresiva e irreversible, siendo la más grave del sistema nervioso central. Ataca sólo a las neuronas motoras. Se desconoce su origen y por ahora no tiene ningún tratamiento curativo, no es contagiosa.
La frecuencia es de 2 por cada 100.000 habitantes
Según avanza la enfermedad, se van atrofiando y paralizando de forma lenta los músculos del cuerpo (movimiento de brazos, piernas, control del tronco y cuello) se va perdiendo la capacidad de hablar, tragar y de respirar.
No daña órganos ni afecta la capacidad intelectual ni los sentidos. El conocido científico británico Stephen Hawking también la padece y solo mueve sus ojos.

Tengo 48 años, casado, con dos hijos, un varón de 17 años y una niña de 14 años, vivo en Eldorado, Misiones, Argentina, trabajo como Bioquímico, aunque a partir del 1 de enero del 2006 deje de trabajar porque me es muy penoso caminar, las escaleras son casi insalvables, y la mano izquierda responde cada vez menos.

Los primeros síntomas fueron en abril de 2005 (aunque llevaba algún tiempo antes notándolo) empecé con calambres, cansancio, dolores musculares… notaba que cada día empeoraba más. Llegaron las caídas sin poder controlar las piernas ,¡¡ al suelo !!, tuve muchas caídas , el miedo se apoderó de mí.
El primer Dr. que me dijo: tiene pinta de esclerosis Lateral Amioatrofica fue D. Rito Quintana, Neurólogo de Posadas Misiones, Entre pruebas y Médicos pasó el tiempo; estaba desesperado…cada vez estaba peor y más asustado,
El día del diagnóstico de certeza (principios de junio de 2005), que me lo hizo el Dr. Dubrovsky Alberto , Neurólogo en Buenos Aires,( profesional de gran tacto y calidad humana), fue el peor de mi vida, me sentía hundido, vacío por dentro, inútil, impotente……Mi cabeza daba vueltas siempre a lo mismo: soy una carga, ¿porqué a mí?, ¿porqué me envían éste castigo?, ¿veré crecer a mis hijos?, ¿cuánto me queda?, ¿como voy a poder con esto?
Conducir es cada vez más difícil, los planes, sueños e ilusiones que tienes se esfuman.
Algunos familiares y amigos se olvidan de que existes, pero no importa, tienes amigos/as que te demuestran que lo son, conoces nuevas amigas que te visitan y se preocupan por ti, en Eldorado me lo han demostrado y lo agradezco mucho, porque veo que todavía hay personas de buen corazón que te quieren por como eres, no por tu enfermedad,. GRACIAS POR la AYUDA Y EL CARIÑO QUE ME DAN.
Ahora estoy mejor, me e dado cuenta que debo seguir para adelante disfrutando cada segundo de mi vida al máximo y valorando todo lo que me rodea. Intento ayudar a nuevos afectados, que están asustados, a vivir el presente y no pensar en el futuro, ¡¡ así me siento útil !!.
ESCLEROSIS
LATERAL
AMIOTRÓFICA
CON ESTE FOLLETO QUIERO QUE CONOZCAN ESTA ENFERMEDAD Y COMO HA CAMBIADO MI VIDA; A MI ESPOSA QUE VA SUPERÁNDOLO POCO A POCO Y A MIS HIJOS QUE SON MUY VALIENTES; LOS QUIERO MUCHO.
Me gustaría conocer afectados de ELA en Argentina , si conocen a alguien y quiere conversar: Tel 03751-421466 o 03751-15666922 / e- mail : eduabauer@yahoo.com.ar o eduardo_bauernfeind@hotmail.com
martes, mayo 16, 2006
Hemos superado las 100 visitas diarias
Tenemos el agrado de comunicarles que hemos superado las 100 visitas diarias. En el día de ayer, 15 de Mayo, recibimos 115 ingresos a nuestro weblog "Esclerosis Lateral Amiotrófica - ELA Argentina".
Desde el 31 de Marzo de 2006 hemos recibido 1306 internautas.
No nos queda mas que agradecer tanto cariño recibido.
Cordialmente.
Carlos Fernández
Desde el 31 de Marzo de 2006 hemos recibido 1306 internautas.
No nos queda mas que agradecer tanto cariño recibido.
Cordialmente.
Carlos Fernández
Autorizan la extracción de células madre de embriones
Ciencia/Salud
Martes 4 de Abril de 2006
En Suiza
Autorizan la extracción de células madre de embriones
BERNA y GINEBRA (EFE).- Por primera vez, Suiza autorizó a un grupo de científicos de la Universidad de Ginebra a realizar una extracción de células madre de embriones humanos, según informaron ayer las autoridades sanitarias del país.
La autorización sólo es para fines científicos, según aseguró la vocera de la Oficina Federal de Salud Pública, quien dijo que Suiza legalizó en 2004 la investigación con células madre de embriones fecundados artificialmente, aunque los científicos usaban células importadas de los Estados Unidos, Suecia e Israel. Con la primera autorización otorgada por las autoridades suizas, los científicos podrán extraer esas células madre, y para eso tendrán a su disposición una reserva, que existe en Suiza, de cien embriones fecundados artificialmente, que fueron obtenidos mediante fecundación in vitro. Esos embriones están destinados a la destrucción, ya que no pueden ser implantados por los posibles defectos genéticos que podrían contener.
Exito terapéutico
En tanto, investigadores suizos y alemanes consiguieron por primera vez en el mundo tratar una afección sanguínea congénita con terapia genética. El ensayo, cuyos resultados publica la revista Nature Medicine, se realizó hace un año y medio en dos pacientes de 25 y 26 años con granulomatosis séptica, una deficiencia del sistema inmune.
Expertos del Hospital Pediátrico Universitario de Zurich y dos colegas de Francfort extrajeron células madre de la sangre de los enfermos y las dotaron de una copia sana del gen defectuoso antes de trasplantarlas en la médula ósea de los jóvenes. Los expertos cobijan cierta cautela, ya que el riesgo de leucemia perdura y no se conoce cuánto demorará esa respuesta terapéutica.
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=794468
LA NACION | 04.04.2006 | Página 11 | Ciencia/Salud
Copyright 2006 SA LA NACION | Todos los derechos reservados
Fuente:
http://www.lanacion.com.ar/
Martes 4 de Abril de 2006
En Suiza
Autorizan la extracción de células madre de embriones
BERNA y GINEBRA (EFE).- Por primera vez, Suiza autorizó a un grupo de científicos de la Universidad de Ginebra a realizar una extracción de células madre de embriones humanos, según informaron ayer las autoridades sanitarias del país.
La autorización sólo es para fines científicos, según aseguró la vocera de la Oficina Federal de Salud Pública, quien dijo que Suiza legalizó en 2004 la investigación con células madre de embriones fecundados artificialmente, aunque los científicos usaban células importadas de los Estados Unidos, Suecia e Israel. Con la primera autorización otorgada por las autoridades suizas, los científicos podrán extraer esas células madre, y para eso tendrán a su disposición una reserva, que existe en Suiza, de cien embriones fecundados artificialmente, que fueron obtenidos mediante fecundación in vitro. Esos embriones están destinados a la destrucción, ya que no pueden ser implantados por los posibles defectos genéticos que podrían contener.
Exito terapéutico
En tanto, investigadores suizos y alemanes consiguieron por primera vez en el mundo tratar una afección sanguínea congénita con terapia genética. El ensayo, cuyos resultados publica la revista Nature Medicine, se realizó hace un año y medio en dos pacientes de 25 y 26 años con granulomatosis séptica, una deficiencia del sistema inmune.
Expertos del Hospital Pediátrico Universitario de Zurich y dos colegas de Francfort extrajeron células madre de la sangre de los enfermos y las dotaron de una copia sana del gen defectuoso antes de trasplantarlas en la médula ósea de los jóvenes. Los expertos cobijan cierta cautela, ya que el riesgo de leucemia perdura y no se conoce cuánto demorará esa respuesta terapéutica.
http://www.lanacion.com.ar/cienciasalud/nota.asp?nota_id=794468
LA NACION | 04.04.2006 | Página 11 | Ciencia/Salud
Copyright 2006 SA LA NACION | Todos los derechos reservados
Fuente:
http://www.lanacion.com.ar/
lunes, mayo 15, 2006
La falta de ayudas bloquea una nueva terapia contra la esclerosis amiotrófica

La falta de ayudas bloquea una nueva terapia contra la esclerosis amiotrófica
Una fundación de Elche promueve un ensayo clínico con células madre de la médula ósea
Fundación Diógenes: info@fundacionela.com - Madrid
EL PAÍS - 09-05-2006
Nieves Berenguer Vicedo, presidenta de la Fundación Diógenes. (JOAQUÍN DE HARO)
En España padecen esclerosis lateral amiotrófica (ELA) 4.000 personas. Se van muriendo poco a poco y otros enfermos los van reemplazando, así que siempre son 4.000, con aproximación matemática asombrosa; 40.000 españoles que hoy están sanos morirán de ELA en los próximos 10 años. Salvo que la ciencia encuentre antes una solución. Esa es la tarea prioritaria de la Fundación Diógenes para la Investigación de la ELA, ubicada en Elche (Alicante). Diez enfermos están preparados para experimentar en ellos un ensayo clínico con terapia celular largamente estudiado. Pero la fundación, superados los obstáculos administrativos, ha tenido que retrasar las investigaciones por falta de dinero para los preceptivos seguros, unos 45.000 euros. Así durante más de un año.
Finalmente, llegó la buena noticia. "Gracias a las asociaciones de ELA, la Cofradía de Bomberos de la Diputación de Córdoba y familiares de afectados, ya casi hemos conseguido recaudar los fondos necesarios para sufragar los gastos de tasas y seguros después de más de dos meses realizando una fuerte campaña divulgativa", dijo el viernes pasado a EL PAÍS una portavoz de la fundación. Los bomberos de Córdoba han donado 24.000 euros. Esperan que los otros 16.000 no tarden en llegar.
El ensayo clínico que se proponen llevar a cabo los especialistas movilizados por la Fundación Diógenes es pionero en España. Lo saben los diez enfermos de ELA que están preparados, e impacientes, para las operaciones previstas. Es su única esperanza.
La ELA es una enfermedad neurodegenerativa que atrofia y paraliza los músculos del cuerpo hasta dejarlos inertes. El avance de este tipo de esclerosis es brutalmente destructivo, desde que el enfermo se da cuenta un día de que la zancada de una de sus piernas se ha acortado, que pierde fuerza en una mano, que le ha cambiado la forma de hablar, o que tiene movimientos musculares anormales -espasmos, sacudidas, calambres o debilidad.
La esperanza de vida de estos enfermos es corta. El afectado dejará de andar, de hablar, de comer, de respirar..., mientras su capacidad intelectual se mantiene intacta. Va a ser consciente siempre de lo que le está pasando. No se conocen sus causas, no hay tratamiento para frenarla, nada puede curarla de momento.
La Fundación Diógenes lleva cinco años en el empeño de sacar adelante la investigación, en colaboración con el Instituto de Neurociencias del Consejo Superior de Investigaciones Científicas (CSIC) de la Universidad Miguel Hernández de Elche y con el apoyo económico del Ayuntamiento de esa ciudad. Su línea de investigación es única en España y la segunda en toda la UE. Fruto de sus trabajos, realizados durante cuatro años, "se ha podido llegar a saber que las células madre de la médula ósea autólogas (del mismo afectado) pueden ser utilizadas de forma segura y eficaz para tratar esta enfermedad neurodegenerativa", dice el informe.
En un modelo animal el trasplante de células madre de la médula ósea en la médula espinal enferma ha producido ya los siguientes beneficios:
1. Las células de la médula ósea son capaces de acomodarse y vivir dentro del sistema nervioso durante un tiempo prolongado
2. Las células de la médula ósea trasplantadas producen un factor beneficioso para las motoneuronas: la molécula llamada GDNF. Esta molécula estimula el desarrollo neuronal y guía a los axones de las células en su crecimiento. Su factor trófico hace que algunas motoneuronas no degeneren, manteniendo en mejores condiciones la actividad motora, rescatándolas de su degeneración, y evitando, por tanto, la progresión de la enfermedad.
Basándose en estos factores, la Fundación Diógenes propuso a mediados del 2005 una investigación denominada Ensayo clínico en fase I/II de utilización de células madre de médula ósea autólogas en pacientes con esclerosis lateral amiotrófica. Su objetivo: "Demostrar la detención de la degeneración de nuevas motoneuronas".
En España padecen ELA 4.000 personas, en buena parte agrupadas en la Asociación de Esclerosis Lateral Amiotrófica (ADELA). Repetidas veces esta organización ha lamentado la falta de atención de las autoridades sanitarias. Su tesis para insistir en un mayor y especial apoyo a los investigadores es que, aunque la causa de la ELA es un misterio, el conocimiento que se tiene del funcionamiento del sistema nervioso es cada vez mayor gracias a los avances de la biología molecular, la ingeniería genética y la bioquímica. Ello permite tener la esperanza de descubrimientos próximos para afrontar la curación de esta rara enfermedad.
Fe de Errores
Los responsables del ensayo clínico promovido por la Fundación Diógenes sobre la esclerosis lateral amiotrófica (ELA) aún no han seleccionado a las personas que van a intervenir, en contra de lo afirmado en la segunda página de Salud del pasado martes. Ello ocurrirá una vez que el experimento cuente con la autorización definitiva de la Agencia Nacional del Medicamento.
Fuente:
http://www.elpais.es/
jueves, mayo 11, 2006
Consultas de Neumonología en pacientes con ELA
C.A.L.Mo.
Notas sobre la Conferencia del Dr. Eduardo Luis De Vito (médico neumonólogo) realizada en C.A.L.Mo., el día 20 de septiembre de 2002.
El Doctor desempeña sus tareas en el Laboratorio Pulmonar, y en la Evaluación de pacientes neurológicos con compromiso de los músculos respiratorios, en el Instituto de Investigaciones Médicas Alfredo Lanari, organismo dependiente de la Universidad de Buenos Aires.
Tema: Consultas de Neumonología en pacientes con E.L.A.
COMO ACTUAR EN CASOS DE AHOGOS
* El principal problema respiratorio que produce la E.L.A en los pacientes, es la DEBILIDAD EN LOS MUSCULOS RESPIRATORIOS. La misma debilidad que los pacientes pueden tener en las piernas, brazos y cuerpo, también se manifiesta en los músculos respiratorios del tórax, el principal de ellos es el diafragma. La forma en que se manifiesta es muy variable, produciendo, en algún momento, sensación de fatiga, ahogos o falta de aire o bien tos débil.
* Una persona puede tener fatiga DISNEA (sensación de dificultad para respirar o de falta de aire) por causas distintas a la debilidad de los músculos del tórax, por ejemplo: bronquitis crónica, hábito de fumar, enfisema, asma, u otras enfermedades. Ninguna de estas causas de fatiga tienen que ver con la E.L.A sino con otra enfermedad preexistente.
* También la falta de aire, puede tener que ver con estados emocionales: problemas de ansiedad, expectativas, crisis de pánico, etc. Estas condiciones deben ser diagnosticadas y tratadas desde el punto de vista psicológico, y con tratamiento farmacológico. En el caso de crisis de pánico o ansiedad, existe una variedad de fármacos, que pueden ser usados en algunos pacientes con E.L.A.
* En todos los casos, siempre hay que tener, para alguna emergencia: tubo de oxígeno, bronco-dilatadores, nebulizador, algún aspirador de secreciones y un kinesiologo cercano.
ESQUEMA DE PAUTAS PARA ESTAR ALERTA
* DEBILIDAD EN LOS MÚSCULOS RESPIRATORIOS
1. MUSCULOS INSPIRATORIOS: el más importante es el diafragma. La debilidad de estos músculos produce DISNEA (fatiga, falta de aire)
2. MUSCULOS ESPIRATORIOS: los más importantes son los abdominales. La debilidad de estos músculos produce ALTERACIONES EN LA CAPACIDAD PARA TOSER, el paciente tiene poca fuerza para toser. La tos es el principal mecanismo de defensa que tiene el organismo para evitar acumulación de secreciones y las infecciones consecuentes. Una tos "mala" produce retención de secreciones e infecciones en los bronquios (bronquitis) o en los pulmones (neumonía).
La tos se compone de las siguientes etapas:
1. INSPIRACION (a cargo de los músculos inspiratorios)
2. CIERRE DE LA GARGANTA (cierre de la glotis para generar presión).
3. AUMENTO DE LA PRESION ABDOMINAL (a cargo de los músculos espiratorios)
4. APERTURA DE LA GLOTIS para toser
En esta enfermedad, hay debilidad de los músculos inspiratorios, la primera etapa está comprometida (1). Algunos pacientes pueden cerrar bien la glotis, y otros, no. Si no cierran bien la glotis, no se genera buena presión (2). Si los músculos espiratorios funcionan mal (3) no hay suficiente presión para desplazar hasta la boca a las secreciones pulmonares.
Las maniobras kinésicas tienden a reemplazar estos mecanismos alterados. Así, las maniobras kinesiológicas con insuflación mediante el Ambú sirven para mejorar estas etapas. El aparato de BiPAP por vía nasal a presiones altas o superiores que las indicadas durante la noche, permite llenar los pulmones con más aire que el que pueden los músculos inspiratorios y así la fuerza para expeler el aire sacarlo se ve aumentada.
Con el problema del cierre de la glotis, no hay muchas maniobras para hacer.
La maniobra de "APRETAR LA PANZA" o sea suplantar la presión que deberían hacer los músculos abdominales, permite la expulsión de secreciones en forma efectiva.
El uso correcto y combinado de todas estas maniobras, disminuye la necesidad de aspiraciones con sondas.
* ALTERACIONES DE LA DEGLUCION
Se denomina DISFAGIA a la INCAPACIDAD PARA TRAGAR BIEN. Generalmente se empieza por tener problemas con los líquidos, semi-sólidos. Rara vez se comienza con los sólidos.
Cuando la alteración de la deglución es muy importante, uno de los problemas es la PERDIDA DE PESO, lo que trae como consecuencia mayor debilidad. Es muy importante evitar la pérdida de peso, para no sumar la debilidad ocasionada por la E.L.A a la ocasionada por la pérdida de peso, ya que ésta es por pérdida de músculo y no sólo de grasa.
Si el paciente puede tragar:
* Maximizar la ingesta de calorías, con dietas hiperproteicas, con proteínas de todo tipo, procesado con puré, para lograr una cohesión de la comida y que no se deshaga en la boca sino que forme una pasta o bolo.
* Cambiar la consistencia de la comida para que sea fácilmente comible.
* Dar suplementos alimentarios como ENSURE PLUS, PULMO CARE, etc.
* Usar espesantes de líquidos, para lograr la consistencia de yoghurt que es más fácil de tragar.
Todo esto apunta a que las alteraciones de la deglución que sufre el paciente, no le produzcan tos y no pierda peso.
Si la enfermedad avanzó o estos recaudos no son suficientes:
* Sonda nasogástrica
* Gastrostomía (más recomendable, más cómoda y más práctica, permite mejor alimentación y no se tapa)
* MANEJO DE LA SALIVA
Con la sonda nasogástrica o la gastrostomía, no se evita el peligro de aspiración en un 100 % ya que también está en juego la secreción de saliva. El ser humano produce alrededor de 1 a 1.5 litros por día de saliva, que se traga naturalmente. Al eliminar el estímulo de la comida, que genera la saliva, ésta disminuye pero no desaparece. El paciente se puede aspirar con la saliva. Existe medicación para disminuir la producción de saliva: papaya, atropina, algunos antidepresivos (bajo prescripción médica), paratropina, etc.
CIGARRILLO Y E.L.A.
* A un paciente con E.L.A. fumador, de 2 o 3 cigarrillos por día, con buena reserva funcional, que no tiene obstrucción bronquial ni asma ni EPOC (enfermedad pulmonar obstructiva crónica), no se le puede negar el fumar. Se trata de buscar el confort del paciente, bajo ciertas reservas. Se puede permitir, siempre que no tenga mala oxigenación en la sangre o esté muy imposibilitado.
LA GASTROSTOMIA
* Existen dos tipos de gastrostomías:
* QUIRÚRGICA: en el quirófano, se hace una incisión de 5 cm a la altura del estómago, con anestesia, se hace un agujerito en el estómago, se pone la cánula y se cierra.
* PERCUTÁNEA: es como una endoscopía. Requiere un set especial de gastrostomía. No necesita anestesia.
No hay que retardar la gastrostomía, si se ha decidido hacerla. Si se hace cuando la capacidad pulmonar todavía está bien y el paciente tiene buen peso, prolonga la sobrevida, porque está mejor alimentado, más cuidado y se evitan los episodios de aspiración. El uso de la gastrostomía no impide que el paciente se alimente por boca. Sólo asegura un ingreso correcto de calorías al estómago para que no pierda peso. También FRENA LA PRESIÓN PSICOLÓGICA sobre el paciente y los familiares respecto de la ingesta de comida.
Caber recordar que si se hace gastrostomía cuando la capacidad pulmonar es superior a la 50%, en esos pacientes, está científicamente demostrado que mejora la sobrevida: están mejor alimentados, mejor cuidados y no tienen episodios de aspiración. En caso contrario, cuando este tiempo ya pasó, se plantea la pregunta: ¿se hace o no se hace la gastrostomía? Si hay mucha presión familiar y del paciente al momento de comer, es adecuado hacerla, esto seguramente va a mejorar la sobrevida sino para mejorar la calidad de vida.
LA TRAQUEOSTOMIA
* En el pasado, se utilizaba con mucha frecuencia si la evolución de la enfermedad así lo requería. En la década del 80 comenzó a usarse un respirador con mascara apoyada sobre la nariz. Se usa una mascara nasal conectada a un respirador que manda aire a los pulmones, aumentando la movilidad del tórax, esto ayuda al diafragma y los músculos respiratorios débiles.
LOS RONQUIDOS
* Existen dos tipos de ronquidos, los BENIGNOS y los NO BENIGNOS estos últimos se asocian a apneas o detención de la respiración por 10 segundos o más. Si los ronquidos son nuevos, o sea que no existían en el paciente antes de la enfermedad, sino que aparecieron con la enfermedad, hay que estudiarlos porque pueden estar relacionados con mala oxigenación de la sangre durante la noche. Si la oxigenación es mala puede ser necesario el uso de un BI-PAP.
* La mala oxigenación de la sangre produce un mal dormir durante la noche, que se traduce en sueño durante el día. Hay que averiguar si el paciente tiene dolor de cabeza a la mañana al levantarse o si se queda dormido durante el día, después de comer, durante una charla, mirando la TV.
* Se pueden realizar estudios de sueño como la POLISOMNOGRAFIA o la SATUROMETRIA.
CUESTIONES A TENER EN CUENTA
1. EVALUACION NEUROLOGICA (Medico Neurólogo)
DIAGNOSTICO Y SEGUIMIENTO
2. EVALUACION RESPIRATORIA (Médico Neumonólogo)
DIAGNOSTICO Y SEGUIMIENTO EN FUNCION DEL DETERIORO GENERAL
3. EVALUACION NUTRICIONAL (Médico Nutricionista)
PARA EVITAR LA PERDIDA DE PESO
4. EVALUACION KINESIOLOGICA (Médico Kinesiólogo)
PARA MIEMBROS SUPERIORES E INFERIORES
PARA MUSCULOS RESPIRATORIOS Y USO DE APARATOS ESPECIFICOS
5. EVALUACION FONOAUDIOLOGICA (Médico Fonoaudiólogo)
FONACION Y DEGLUCION
MANIOBRAS, TIPO DE MASTICACION, FONACION, HABLA.
6. AYUDAS A LA RESPIRACION
DECISION COMPARTIDA CON EL PACIENTE-FAMILIA-MEDICO
TRAQUEOSTOMIA O BI-PAP Y USO DE OXIGENO EN ALGUNOS CASOS SELECCIONADOS
Fuente:
http://www.calmo.org.ar/
Notas sobre la Conferencia del Dr. Eduardo Luis De Vito (médico neumonólogo) realizada en C.A.L.Mo., el día 20 de septiembre de 2002.
El Doctor desempeña sus tareas en el Laboratorio Pulmonar, y en la Evaluación de pacientes neurológicos con compromiso de los músculos respiratorios, en el Instituto de Investigaciones Médicas Alfredo Lanari, organismo dependiente de la Universidad de Buenos Aires.
Tema: Consultas de Neumonología en pacientes con E.L.A.
COMO ACTUAR EN CASOS DE AHOGOS
* El principal problema respiratorio que produce la E.L.A en los pacientes, es la DEBILIDAD EN LOS MUSCULOS RESPIRATORIOS. La misma debilidad que los pacientes pueden tener en las piernas, brazos y cuerpo, también se manifiesta en los músculos respiratorios del tórax, el principal de ellos es el diafragma. La forma en que se manifiesta es muy variable, produciendo, en algún momento, sensación de fatiga, ahogos o falta de aire o bien tos débil.
* Una persona puede tener fatiga DISNEA (sensación de dificultad para respirar o de falta de aire) por causas distintas a la debilidad de los músculos del tórax, por ejemplo: bronquitis crónica, hábito de fumar, enfisema, asma, u otras enfermedades. Ninguna de estas causas de fatiga tienen que ver con la E.L.A sino con otra enfermedad preexistente.
* También la falta de aire, puede tener que ver con estados emocionales: problemas de ansiedad, expectativas, crisis de pánico, etc. Estas condiciones deben ser diagnosticadas y tratadas desde el punto de vista psicológico, y con tratamiento farmacológico. En el caso de crisis de pánico o ansiedad, existe una variedad de fármacos, que pueden ser usados en algunos pacientes con E.L.A.
* En todos los casos, siempre hay que tener, para alguna emergencia: tubo de oxígeno, bronco-dilatadores, nebulizador, algún aspirador de secreciones y un kinesiologo cercano.
ESQUEMA DE PAUTAS PARA ESTAR ALERTA
* DEBILIDAD EN LOS MÚSCULOS RESPIRATORIOS
1. MUSCULOS INSPIRATORIOS: el más importante es el diafragma. La debilidad de estos músculos produce DISNEA (fatiga, falta de aire)
2. MUSCULOS ESPIRATORIOS: los más importantes son los abdominales. La debilidad de estos músculos produce ALTERACIONES EN LA CAPACIDAD PARA TOSER, el paciente tiene poca fuerza para toser. La tos es el principal mecanismo de defensa que tiene el organismo para evitar acumulación de secreciones y las infecciones consecuentes. Una tos "mala" produce retención de secreciones e infecciones en los bronquios (bronquitis) o en los pulmones (neumonía).
La tos se compone de las siguientes etapas:
1. INSPIRACION (a cargo de los músculos inspiratorios)
2. CIERRE DE LA GARGANTA (cierre de la glotis para generar presión).
3. AUMENTO DE LA PRESION ABDOMINAL (a cargo de los músculos espiratorios)
4. APERTURA DE LA GLOTIS para toser
En esta enfermedad, hay debilidad de los músculos inspiratorios, la primera etapa está comprometida (1). Algunos pacientes pueden cerrar bien la glotis, y otros, no. Si no cierran bien la glotis, no se genera buena presión (2). Si los músculos espiratorios funcionan mal (3) no hay suficiente presión para desplazar hasta la boca a las secreciones pulmonares.
Las maniobras kinésicas tienden a reemplazar estos mecanismos alterados. Así, las maniobras kinesiológicas con insuflación mediante el Ambú sirven para mejorar estas etapas. El aparato de BiPAP por vía nasal a presiones altas o superiores que las indicadas durante la noche, permite llenar los pulmones con más aire que el que pueden los músculos inspiratorios y así la fuerza para expeler el aire sacarlo se ve aumentada.
Con el problema del cierre de la glotis, no hay muchas maniobras para hacer.
La maniobra de "APRETAR LA PANZA" o sea suplantar la presión que deberían hacer los músculos abdominales, permite la expulsión de secreciones en forma efectiva.
El uso correcto y combinado de todas estas maniobras, disminuye la necesidad de aspiraciones con sondas.
* ALTERACIONES DE LA DEGLUCION
Se denomina DISFAGIA a la INCAPACIDAD PARA TRAGAR BIEN. Generalmente se empieza por tener problemas con los líquidos, semi-sólidos. Rara vez se comienza con los sólidos.
Cuando la alteración de la deglución es muy importante, uno de los problemas es la PERDIDA DE PESO, lo que trae como consecuencia mayor debilidad. Es muy importante evitar la pérdida de peso, para no sumar la debilidad ocasionada por la E.L.A a la ocasionada por la pérdida de peso, ya que ésta es por pérdida de músculo y no sólo de grasa.
Si el paciente puede tragar:
* Maximizar la ingesta de calorías, con dietas hiperproteicas, con proteínas de todo tipo, procesado con puré, para lograr una cohesión de la comida y que no se deshaga en la boca sino que forme una pasta o bolo.
* Cambiar la consistencia de la comida para que sea fácilmente comible.
* Dar suplementos alimentarios como ENSURE PLUS, PULMO CARE, etc.
* Usar espesantes de líquidos, para lograr la consistencia de yoghurt que es más fácil de tragar.
Todo esto apunta a que las alteraciones de la deglución que sufre el paciente, no le produzcan tos y no pierda peso.
Si la enfermedad avanzó o estos recaudos no son suficientes:
* Sonda nasogástrica
* Gastrostomía (más recomendable, más cómoda y más práctica, permite mejor alimentación y no se tapa)
* MANEJO DE LA SALIVA
Con la sonda nasogástrica o la gastrostomía, no se evita el peligro de aspiración en un 100 % ya que también está en juego la secreción de saliva. El ser humano produce alrededor de 1 a 1.5 litros por día de saliva, que se traga naturalmente. Al eliminar el estímulo de la comida, que genera la saliva, ésta disminuye pero no desaparece. El paciente se puede aspirar con la saliva. Existe medicación para disminuir la producción de saliva: papaya, atropina, algunos antidepresivos (bajo prescripción médica), paratropina, etc.
CIGARRILLO Y E.L.A.
* A un paciente con E.L.A. fumador, de 2 o 3 cigarrillos por día, con buena reserva funcional, que no tiene obstrucción bronquial ni asma ni EPOC (enfermedad pulmonar obstructiva crónica), no se le puede negar el fumar. Se trata de buscar el confort del paciente, bajo ciertas reservas. Se puede permitir, siempre que no tenga mala oxigenación en la sangre o esté muy imposibilitado.
LA GASTROSTOMIA
* Existen dos tipos de gastrostomías:
* QUIRÚRGICA: en el quirófano, se hace una incisión de 5 cm a la altura del estómago, con anestesia, se hace un agujerito en el estómago, se pone la cánula y se cierra.
* PERCUTÁNEA: es como una endoscopía. Requiere un set especial de gastrostomía. No necesita anestesia.
No hay que retardar la gastrostomía, si se ha decidido hacerla. Si se hace cuando la capacidad pulmonar todavía está bien y el paciente tiene buen peso, prolonga la sobrevida, porque está mejor alimentado, más cuidado y se evitan los episodios de aspiración. El uso de la gastrostomía no impide que el paciente se alimente por boca. Sólo asegura un ingreso correcto de calorías al estómago para que no pierda peso. También FRENA LA PRESIÓN PSICOLÓGICA sobre el paciente y los familiares respecto de la ingesta de comida.
Caber recordar que si se hace gastrostomía cuando la capacidad pulmonar es superior a la 50%, en esos pacientes, está científicamente demostrado que mejora la sobrevida: están mejor alimentados, mejor cuidados y no tienen episodios de aspiración. En caso contrario, cuando este tiempo ya pasó, se plantea la pregunta: ¿se hace o no se hace la gastrostomía? Si hay mucha presión familiar y del paciente al momento de comer, es adecuado hacerla, esto seguramente va a mejorar la sobrevida sino para mejorar la calidad de vida.
LA TRAQUEOSTOMIA
* En el pasado, se utilizaba con mucha frecuencia si la evolución de la enfermedad así lo requería. En la década del 80 comenzó a usarse un respirador con mascara apoyada sobre la nariz. Se usa una mascara nasal conectada a un respirador que manda aire a los pulmones, aumentando la movilidad del tórax, esto ayuda al diafragma y los músculos respiratorios débiles.
LOS RONQUIDOS
* Existen dos tipos de ronquidos, los BENIGNOS y los NO BENIGNOS estos últimos se asocian a apneas o detención de la respiración por 10 segundos o más. Si los ronquidos son nuevos, o sea que no existían en el paciente antes de la enfermedad, sino que aparecieron con la enfermedad, hay que estudiarlos porque pueden estar relacionados con mala oxigenación de la sangre durante la noche. Si la oxigenación es mala puede ser necesario el uso de un BI-PAP.
* La mala oxigenación de la sangre produce un mal dormir durante la noche, que se traduce en sueño durante el día. Hay que averiguar si el paciente tiene dolor de cabeza a la mañana al levantarse o si se queda dormido durante el día, después de comer, durante una charla, mirando la TV.
* Se pueden realizar estudios de sueño como la POLISOMNOGRAFIA o la SATUROMETRIA.
CUESTIONES A TENER EN CUENTA
1. EVALUACION NEUROLOGICA (Medico Neurólogo)
DIAGNOSTICO Y SEGUIMIENTO
2. EVALUACION RESPIRATORIA (Médico Neumonólogo)
DIAGNOSTICO Y SEGUIMIENTO EN FUNCION DEL DETERIORO GENERAL
3. EVALUACION NUTRICIONAL (Médico Nutricionista)
PARA EVITAR LA PERDIDA DE PESO
4. EVALUACION KINESIOLOGICA (Médico Kinesiólogo)
PARA MIEMBROS SUPERIORES E INFERIORES
PARA MUSCULOS RESPIRATORIOS Y USO DE APARATOS ESPECIFICOS
5. EVALUACION FONOAUDIOLOGICA (Médico Fonoaudiólogo)
FONACION Y DEGLUCION
MANIOBRAS, TIPO DE MASTICACION, FONACION, HABLA.
6. AYUDAS A LA RESPIRACION
DECISION COMPARTIDA CON EL PACIENTE-FAMILIA-MEDICO
TRAQUEOSTOMIA O BI-PAP Y USO DE OXIGENO EN ALGUNOS CASOS SELECCIONADOS
Fuente:
http://www.calmo.org.ar/
![]()

